
Product Advantages
Ultra-long read length, easy access to full-length transcript sequence, low multi-site comparison rate
Most traditional sequencing methods require fragmentation, and it is still difficult to accurately assemble a complete transcript, especially when a read length can be compared to multiple sites (such as highly conserved sequence segments). The long read length advantage of ONT sequencing allows the easy sequencing of the full-length transcript sequence from the 5′ end to the poly(A) tail, significantly reducing the multi-site comparison of reads and increasing the specificity of the comparison between reads and reference genomes.
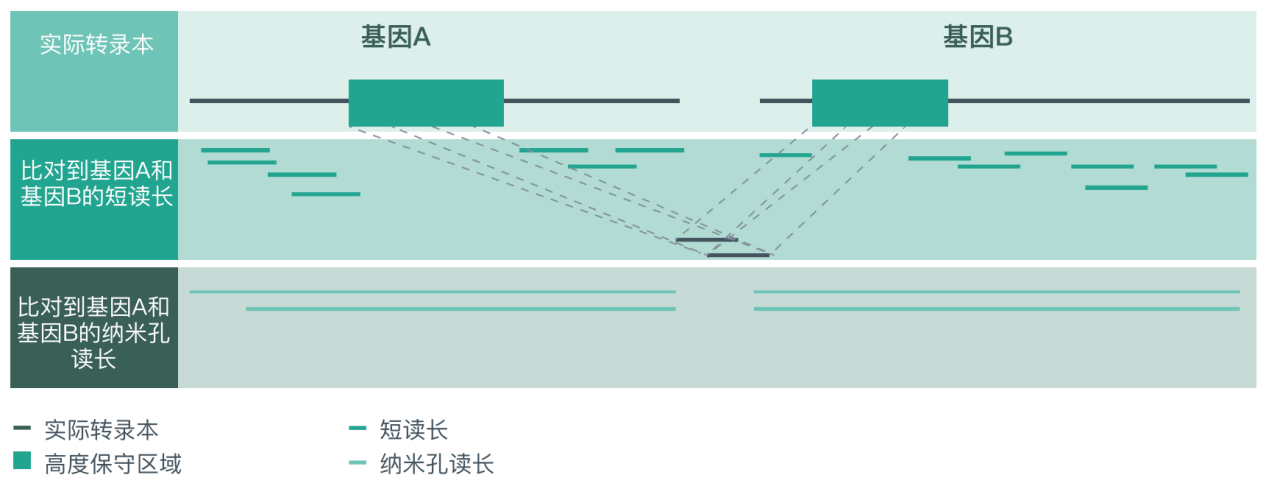
Use short reads and Nanopore long reads to assemble full-length transcripts
Completely cover splicing site,accurately identify alternative splicing events
When eukaryotic genes are transcribed, the process by which an mRNA precursor produces different mRNA splicing isoforms through different splicing methods is called alternative splicing (or alternative splicing, AS). Alternative splicing allows each gene to produce a large number of mRNA isoforms, thereby promoting the diversity of gene composition and gene functions. The short read lengths produced by traditional RNA sequencing technologies lose positional information, making it challenging to assemble the selective splicing mRNA isoforms correctly. The long read length of the nanopore can span the splicing site, thereby accurately identifying variable splicing events and directly performing quantitative analysis of transcript expression.
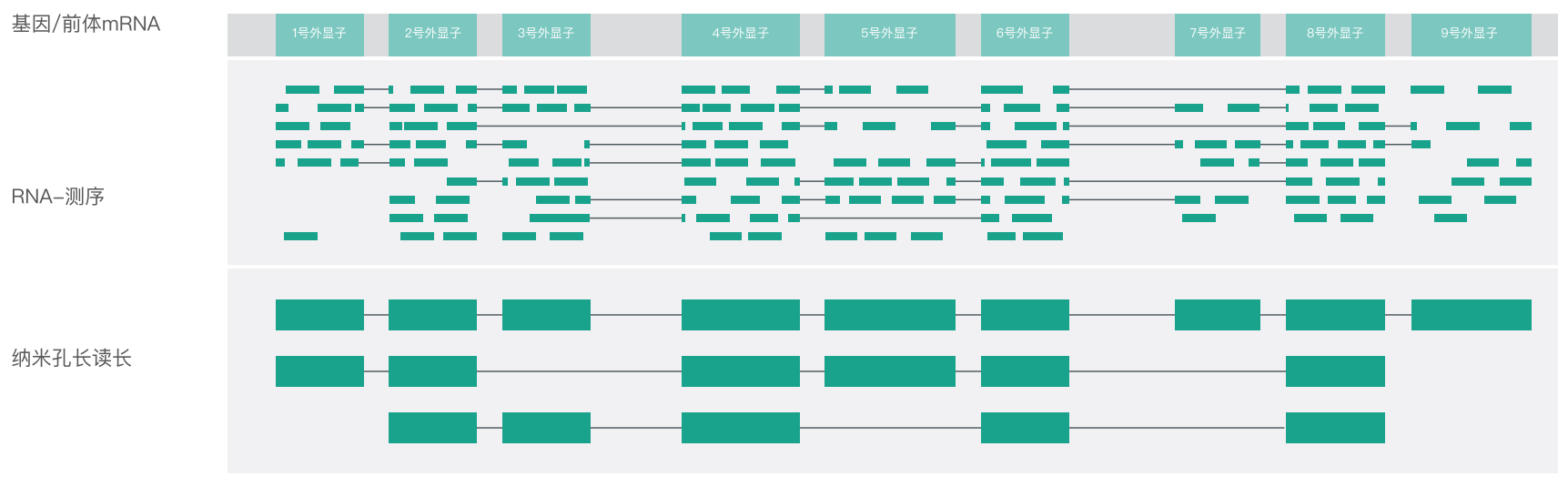
Use short reads and Nanopore long reads to assemble full-length transcripts
Quantitative expression analysis of transcriptional isoforms
Short-read sequencing cannot accurately identify transcript isoforms due to its short read length and high multi-site comparison rate. Therefore, accurate expression results cannot be obtained at the transcript level. The ONT transcriptome sequencing reads are long enough to directly obtain transcriptional isoform sequences. Furthermore, its high throughput easily allows the saturation of sequencing, leading to more accurate expression analysis at both the gene level and the transcript level. ERCC spike-in analysis shows that all types of ONT transcriptome sequencing libraries are quantifiable.
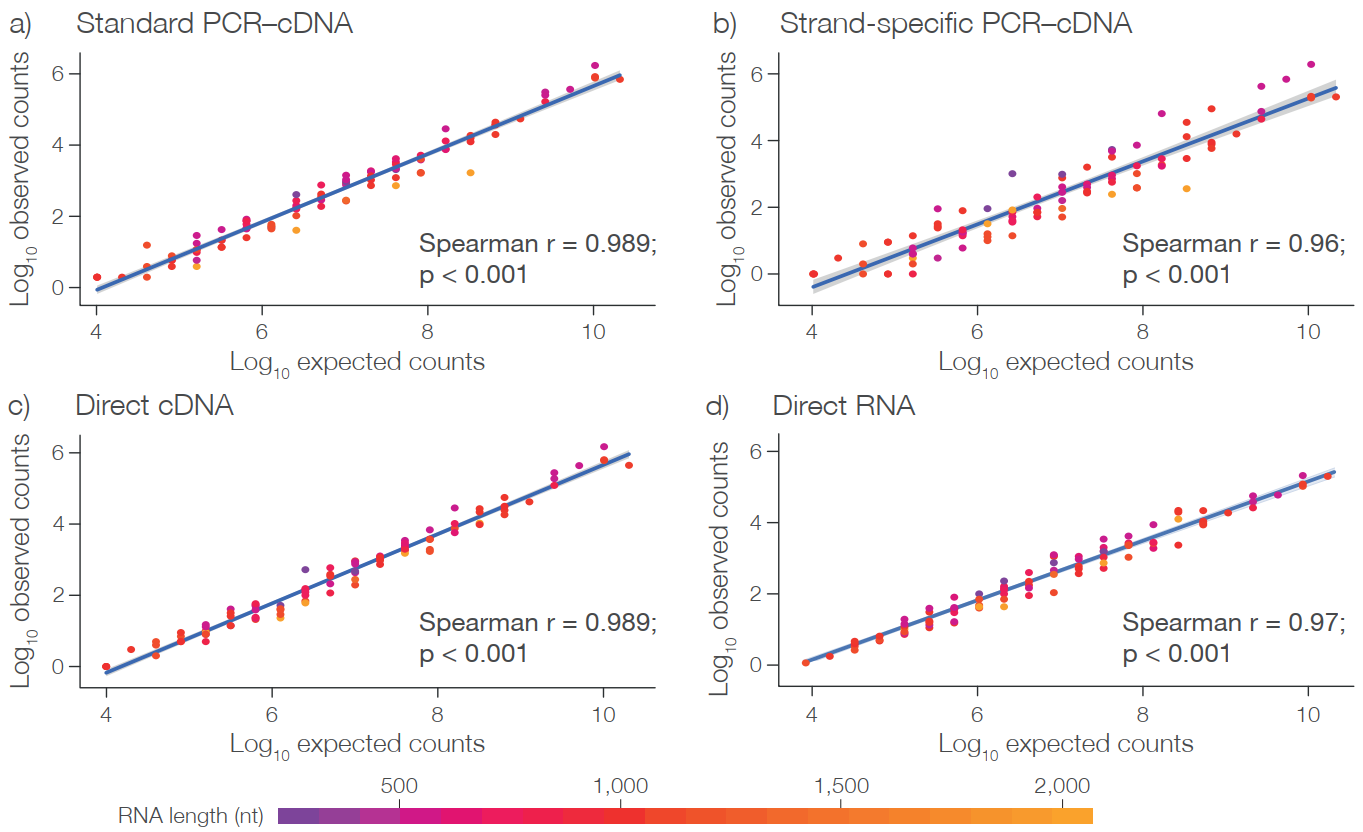
Nanopore full-length transcriptome quantitative analysis
Strategy
| RNA Sample Size | Library Construction Strategy | Sequencing Strategy |
| Total ≥ 0.5 μg, Concentration ≥50 ng/μL | cDNA PCR amplification then 1D library construction | Gene quantification required: ONT sequencing data volume ≥ 2 Gb |
| Total | cDNA PCR amplification then 1D library construction | Gene quantification required: ONT sequencing data volume ≥ 15 Gb |
Analysis
- Raw Data Quality Control
- Full-Length Transcript Identification
- Reference transcriptome comparison
- Gene Function and Transcript Structure Annotation
- Differential gene/transcription isoform quantification
- Function enrichment, protein interaction analysis
Results
Full-length transcript recognition
During the library building process, different primers will be added at both ends of the reads: one end is a 5’primer while the other end is a 3’primer, and there is also a polyA sequence before the 3’primer. If the transcript extracted in the experiment is a full-length transcript, then the complete insert sequence is the full-length transcript sequence. By judging the existence and positional relationship of the 5’primer, the 3’primer, and the polyA, the insert sequence is classified. Full-length reads are the full-length sequences obtained by sequencing.
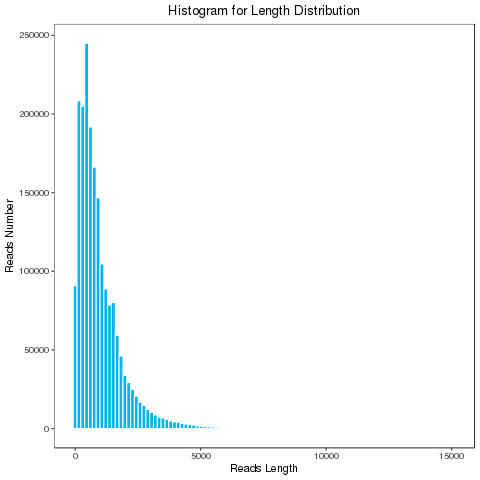
Full Length Sequence Distribution
Fusion gene recognition
Fusion genes are chimeric genes formed by fusion of all or part of the sequences of two genes. It may be formed by a chromosome translocation, an intermediate deletion or a chromosome inversion.
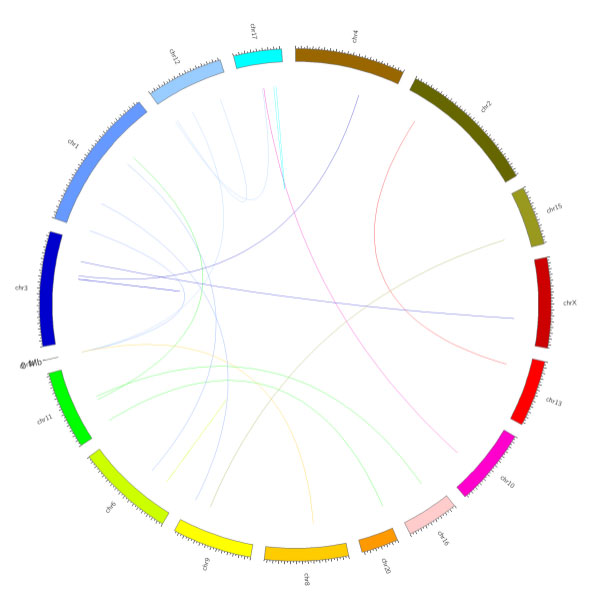
Fusion gene circos map
Transcript clustering to remove redundancy
Since there may be multiple copies of the same transcript at a time point in the cell, and different transcript copies may be degraded to varying degrees at the 5’end during the sequencing process, the full-length reads obtained by sequencing include many redundant sequences derived from the same transcript. It is necessary to cluster full-length sequences to eliminate redundancy.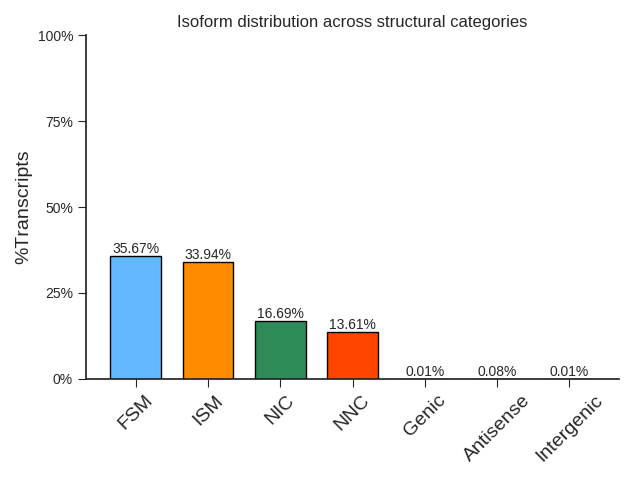
Isoform evaluation results
Alternative Splice
Alternative splice (Alternative Splice) is an important mechanism for regulating gene expression and generating proteome diversity.
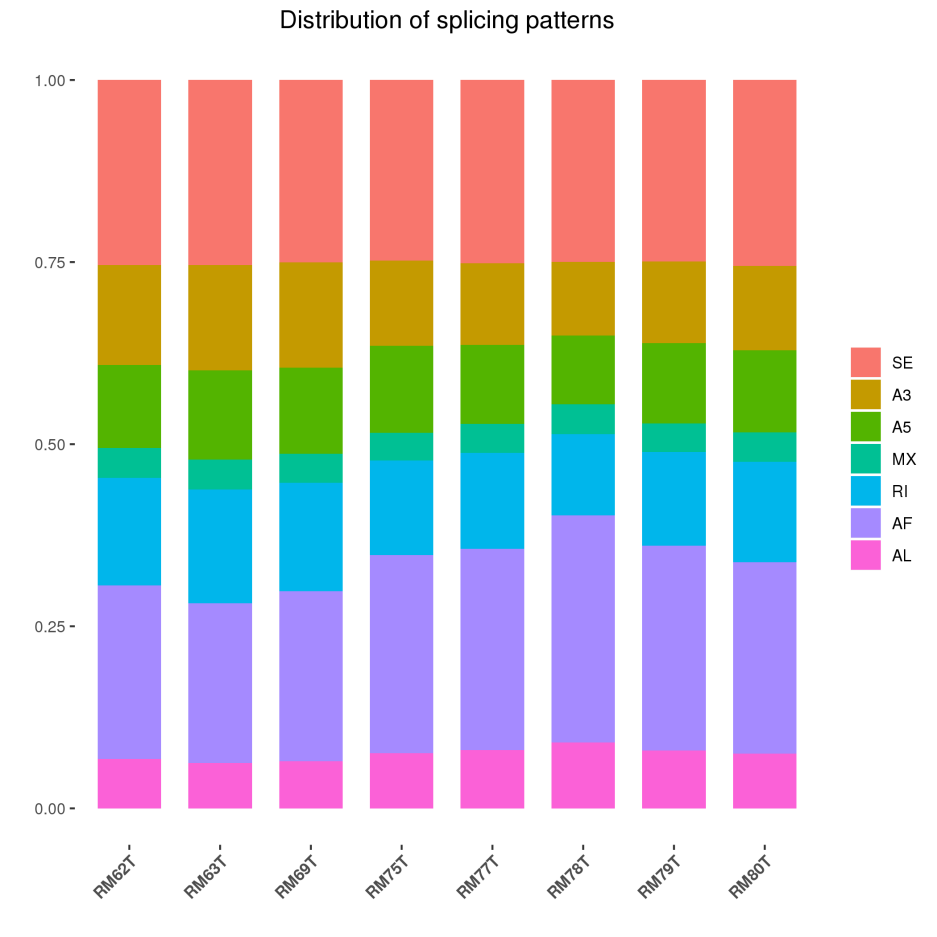
Alternative splicing type distribution
Differential gene clustering
Differential gene cluster analysis is used to judge the clustering mode of differential gene expression under different experimental conditions.
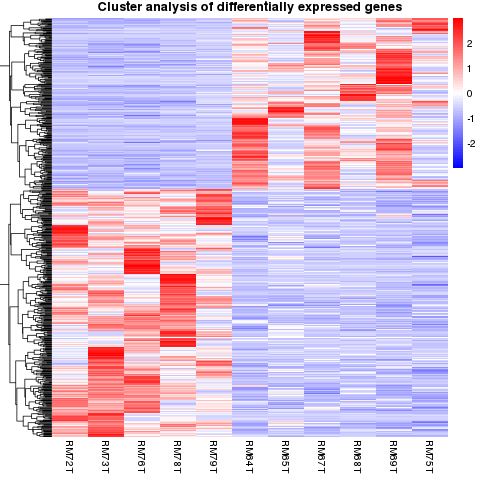
Differential Transcript Expression
In transcriptome analysis, DGE (Differential Gene Expression) refers to the differential expression of genes under different conditions, DTE (Differential Transcript Expression) refers to the differential expression of transcripts (the expression of the gene to which the transcript belongs may change significantly, or there may be no significant change), and DTU (Differential Transcript Usage) refers to when different conditions have not caused change in gene expression, but have resulted in significant change in the expression of the transcript belonging to the gene.
Because Nanopore transcriptome sequencing can be quantified at the transcript level, it can further analyze the differential expression of transcripts under different conditions (Differential Transcript Expression).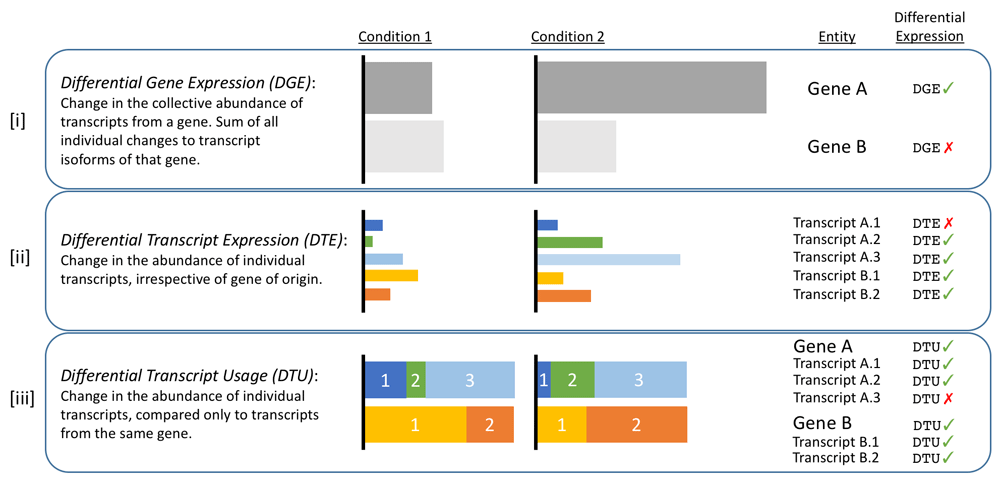
[i] GeneA was differentially expressed in condition1 and condition2, but GeneB was not;
[ii] GeneA transcripts A2 and A3 are differentially expressed, while A1 does not change. GeneB transcripts B1 and B2 are differentially expressed, but the overall expression of GeneB does not change (in this case, B1 and B2 are both DTE and DTU);;
[iii] The expression of GeneA and GeneB did not change under the two conditions, but the expression of transcripts A1, A2 and B1, B2 changed significantly. This belongs to DTU.
Common Questions
1 What are the advantages of ONT PCR cDNA over PacBio iso-Seq?
It can be accurately quantified at both the gene level and the transcript level with a lower cost. Taking human samples as an example, Sequel II requires 2 cells to reach saturation in the sequencing of transcripts, while ONT only requires one cell to reach saturation for the transcripts of 6 samples, and at a lower demand for original sample size.
2 Does ONT PCR cDNA need to be repeated?
According to research needs, the focus is on variable shearing of transcripts. Biological repetitions are not required. If you are concerned about the results of quantitative analysis, you must do repetitions. At least three are recommended.
Case Analysis
Case Analysis
Detection and quantification of cancer-specific transcript variants using ONT full-length transcriptome
SF3B1 is the coding gene of the key factor of RNA splicing body. Mutations in this gene will affect the normal splicing of RNA and cause abnormal expression of downstream genes. Studies have shown that SF3B1 has a higher frequency of mutations in a variety of tumor cells, of which chronic lymphocytic leukemia (CLL) patients are the most prominent. Although the alternative splicing pattern induced by the SF3B1 mutation has been previously analyzed by RNA-Seq, the short read length limits the systematic identification of this pattern at the isoform level. In addition, intron retentions (IR) are very common in various cancers and can distinguish tumors from matched normal tissues. Short-read technology can easily cause misclassification of intron retention events, especially in complex alternative splicing regions, and transcripts containing intron retention cannot be quantitatively analyzed.
The study used nanopore full-length transcriptome data to identify and analyze full-length transcripts associated with SF3B1 mutations in chronic lymphocytic leukemia samples. Nanopore sequencing technology can sequence full-length transcripts, directly obtain full-length isoform, and accurately identify intron retention, and the high-throughput PromethION sequencer can generate a sufficient number of full-length sequence transcripts for cancer-specific transcription variant detection and quantification.
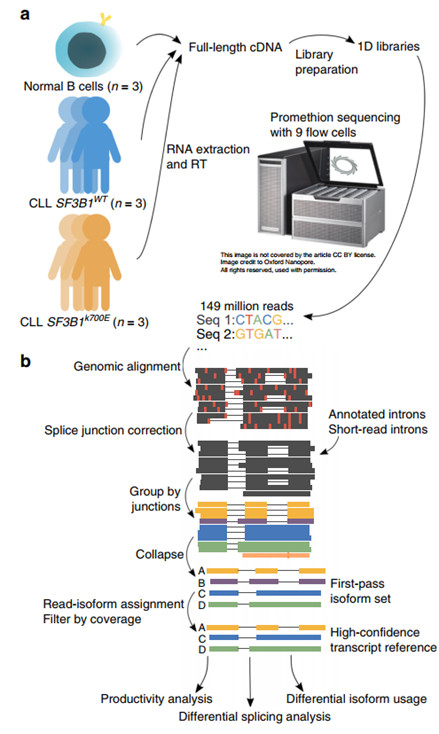
Figure 1 Long-read long nanopore sequencing and FLAIR sequence analysis process
The researchers selected 3 SF3B1 mutation-free CLL patient samples (CLL SF3B1WT), 3 SF3B1K700E mutation CLL patient samples (CLL SF3B1K700E), and 3 ordinary B lymphocyte samples for ONT full-length transcriptome sequencing, obtaining a total of about 257M reads. (Figure 1a). Subsequently, the researchers developed the FLAIR process for identifying high-confidence transcripts. The process includes three main steps: comparison, correction, and integration. Firstly, one must compare the original reads of all samples with the genome to identify the general transcript structure, then use short read sequencing data to correct the mistaken splice junctions in the reads due to sequencing errors, base deletions, and alignment difficulties. Reads with the same splice junction sequence are integrated into the corresponding isoform group, and all the reads are re-aligned to these isoforms. Finally the isoforms supported by less than three reads are filtered out, and the remaining isoforms with sufficient coverage constitute the final high confidence isoform set (Figure 1b).
Using FLAIR, the researchers identified 326,699 high-confidence splicing isoforms, most of which (90.0%) were unannotated isoforms. Of these unannotated isoforms, 142,971 are new combinations formed by annotated splice junctions, and the rest are isoforms with intron retention (21,700) or new exons (3,594). The researchers used the nanopore data to verify the differential changes in the 3’splice site reported by previous researchers related to the SF3B1 mutation. The results show that long reads not only enable the identification of the altered splice sites of mutant SF3B1, but also associate these splice site abnormal events and other similar events with the corresponding full-length isoform.
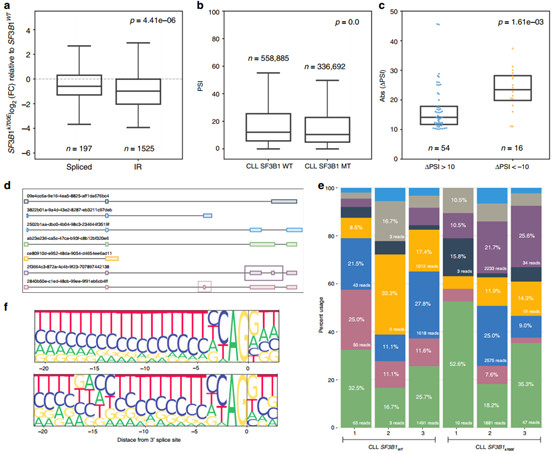
Figure 2 Intron retention events are more strongly down-regulated in CLL SF3B1K700E
Subsequently, the researchers explored the changes in intron retention isoform associated with SF3B1 mutations. By comparing the expression profile differences between CLL samples, they found that the expression of intron retention isoform in SF3B1K700E samples was generally down-regulated compared with SF3B1WT samples (Figure 2a). Further research found that the use of re-analysis of mutant SF3B1 CLL, Nalm-6 cell line and TCGA BRCA short-read data set confirmed this down-regulation (Figure 2d-e).
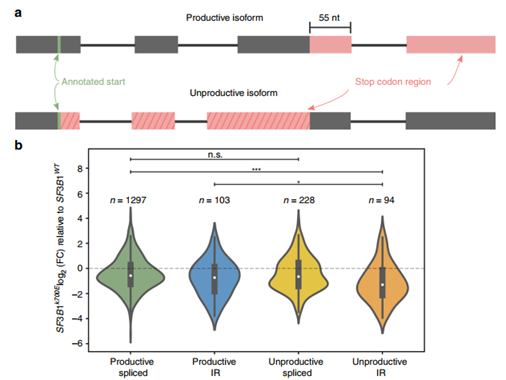
Figure 3 Mutant SF3B1 down-regulates transcripts retained by “invalid” introns
In addition, the study also found that full-length reads can not only improve the recognition of intron-reserved transcripts, but can also better estimate the abundance of valid and invalid isoforms (Figure 3a). The researchers classified the differentially expressed isoforms between the SF3B1K700E and SF3B1WT samples according to whether they were effective and whether they contained intron retention. The results showed that the expression of invalid and intron retention transcripts was down-regulated (Figure 3b). GO analysis found these invalid and intron-retained transcripts are related to the kinase signaling pathway. Researchers speculate that they may be introns trapped in the nucleus, and downregulation of expression may lead to the expression of kinase-related genes, thereby supporting tumor proliferation, but further experimental verification is still needed.
References:Tang, A.D.,Soulette, C.M., van Baren, M.J. etal. Full-length transcript characterization of SF3B1 mutation in chroniclymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11, 1438 (2020). https://doi.org/10.1038/s41467-020-15171-6
 GrandOmics collaborates with Oxford Nanopore to deliver...2019-04-26 - pm2:11
GrandOmics collaborates with Oxford Nanopore to deliver...2019-04-26 - pm2:11 Conference Announcement | January 12-17, GrandOmics sincerely...2024-01-12 - pm3:40
Conference Announcement | January 12-17, GrandOmics sincerely...2024-01-12 - pm3:40 GrandOmics X PacBio Revio – Higher throughput, more...2023-06-09 - pm7:06
GrandOmics X PacBio Revio – Higher throughput, more...2023-06-09 - pm7:06 NextOmics: the FIRST officially certified service provider...2018-01-18 - pm3:00
NextOmics: the FIRST officially certified service provider...2018-01-18 - pm3:00
Follow us on WeChat

GrandOmics

Grandomics Clinical Services

Grandomics Research Services