
Comparison of short read length, long read length and ONT direct RNA sequencing analysis
Product Advandages
Direct RNA sequencing does not require PCR and has no sequencing GC preference
Because the PCR process has GC preference, it is not easy to amplify sequences with too high or too low GC content. Therefore, short-read sequencing will introduce GC preference during database construction and sequencing, which reduces the accuracy of quantitative analysis. ONT sequencing technology (direct cDNA & direct RNA) does not require PCR amplification, providing unbiased, full-length, strand-specific RNA sequences.
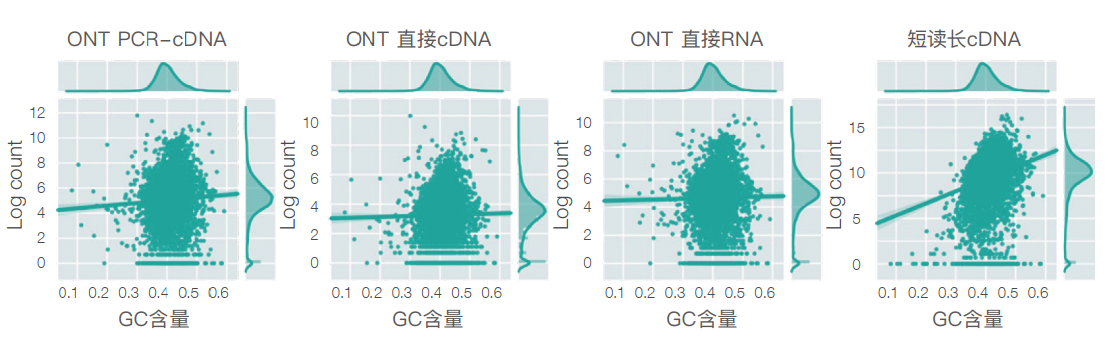
The GC bias in the nanopore data set is lower than that in the short read data set
Accurately detect transcript poly(A) tail length
The poly(A) tail of the transcript is believed to play an important role in post-transcriptional regulation, including mRNA stability and translation efficiency. The length of the poly(A) tail can reach hundreds of nucleotides, and it is difficult to measure such information using short-read sequencing data. The full-length transcript obtained by ONT direct RNA sequencing contains poly(A) tail information. ONT technology allows the use of algorithm tools to calculate the length of the poly(A) tail, the estimation of the poly(A) tail length of each read sequence, and even detection of heterogeneity differences between poly(A) tails of different isoforms.
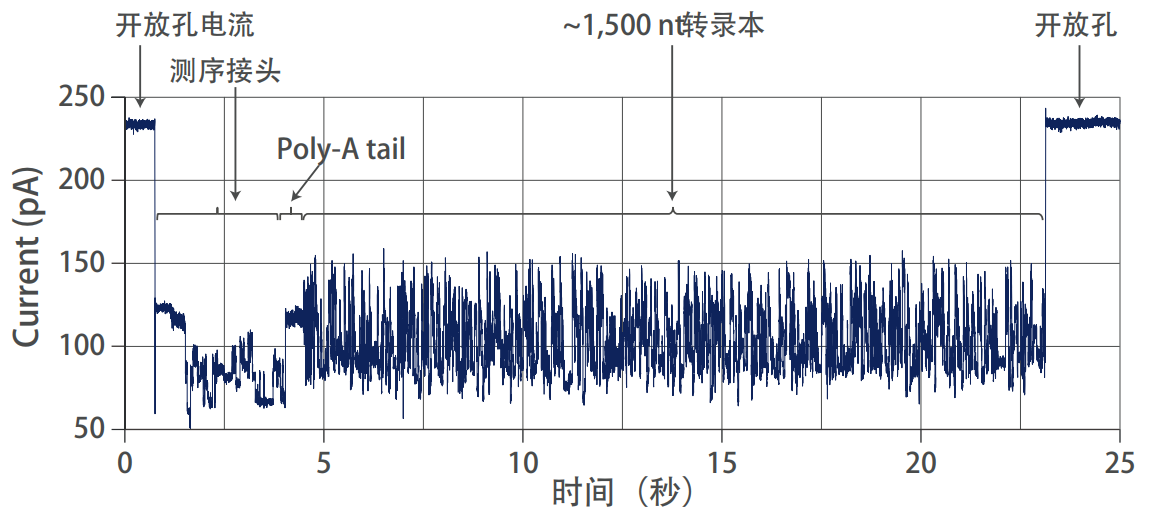
Nanopore direct RNA sequencing to identify transcript poly(A) tail length
Direct RNA sequencing to identify RNA base modification information
Short-read sequencing requires PCR in the process of library construction, and thus loses base modification information in RNA molecules. Direct RNA sequencing does not require amplification or strand synthesis: during the sequencing process, modified bases pass directly through the nanopore, generating a current signal different from unmodified bases in the original signal. Identification of the base modification information is possible through a specific software algorithm.
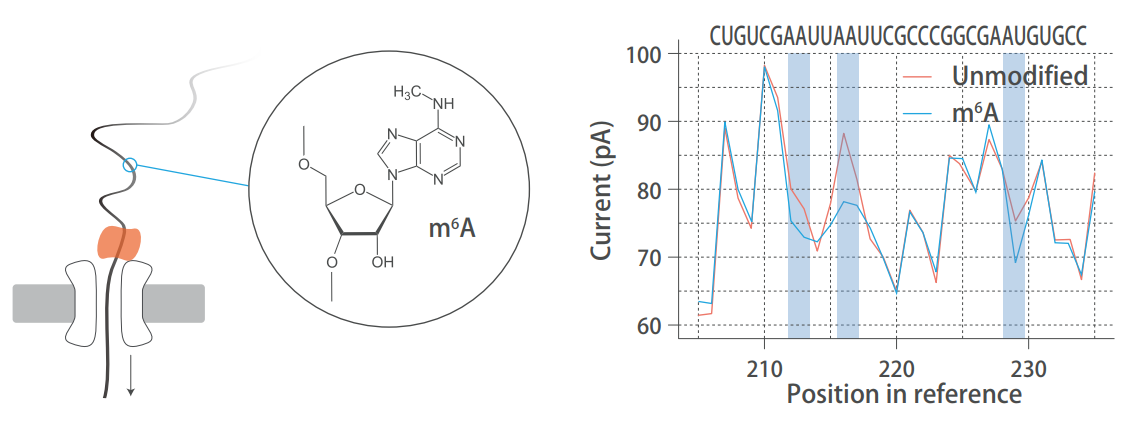
Nanoproe direct RNA sequencing detects natural RNA modifications
Strategy
| RNA Sample | Library Construction Strategy | Sequencing Strategy |
| Total ≥ 50 μg, Concentration ≥180 ng/μL | Direct RNA 1D Library Construction | Depending on specific case |
Analysis
- Quality control of raw data
- RNA base modification detection
- Poly(A) tail length estimation
- Reference transcriptome alignment
- Gene function and transcript structure annotation
- Differential gene/transcription isoform quantification
- Function enrichment, protein interaction analysis
Results
Transcript clustering to remove redundancy
Since there may be multiple copies of the same transcript at a time point in the cell, and different transcript copies may be degraded to varying degrees at the 5’end during the sequencing process, many of the sequenced full-length reads are redundant sequences derived from the same transcript, and the full-length sequences need to be clustered to eliminate redundancy.
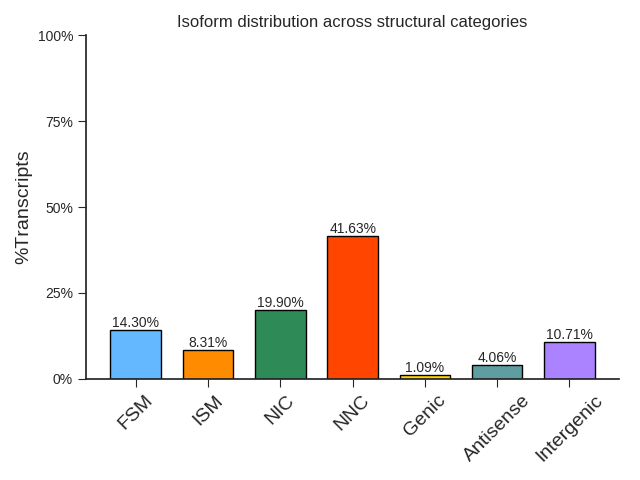
isoform evaluation results
Alternative Splice
Alternative splice (Alternative Splice) is an important mechanism for regulating gene expression and generating proteome diversity.
Alternative splicing identification
Poly(A) tail length estimation
Since the direct-RNA sequencing method sequences from the 3’end to the 5’end of the RNA, the poly(A) information is completely preserved during the process of adding the poly(T) adapter. Thus, an estimate of the RNA poly(A)tail legnth can be read from the electrical signal of the direct-RNA sequencing.
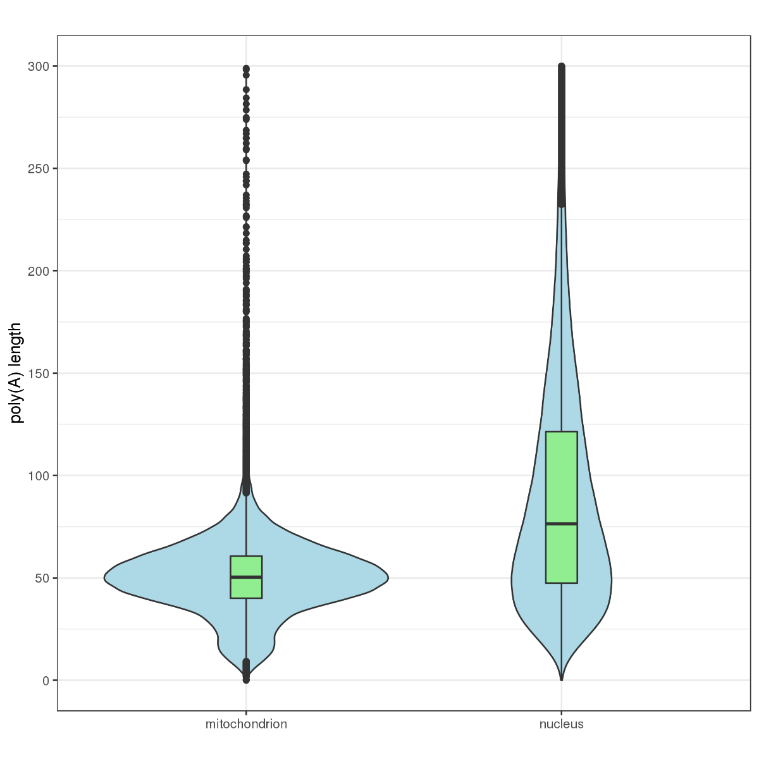
Poly(A) tail length of different genes and transcripts
RNA methylation analysis
Other sequencing methods need reverse transcription or PCR during the sequencing process of the library, causing a loss of base modification information of the RNA molecule. Meanwhile, direct-RNA sequencing does not require reverse transcription or PCR, which means that nucleotides that have underwent modification pass directly through the nanopore, producing different current characteristics from unmodified bases in the original electrical signal. Identification of the base modification information is possible through a specific software algorithm.
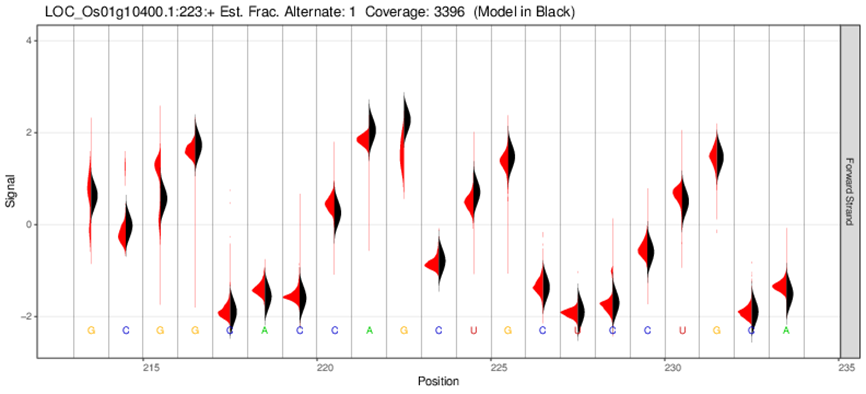
Electrical signal distribution map of methylation sites
Common Questions
- What is the minimum amount of RNA required to build a Direct RNA library?
Qualified total RNA 40-80ug, concentration ≥180 ng/μL.
- 2. What is the approximate output of Direct RNA 1 cell?
Because there is no PCR amplification process for Direct RNA library construction and sequencing, the data volume of PCR cDNA full-length transcriptome is relatively low. We can promise that the amount of high-quality total RNA is not less than 1Gb.
Case Study
Case Study
Nanopore direct RNA sequencing of human poly(A) transcriptome
High-throughput cDNA sequencing has given us a deeper understanding of the complexity and regulation of the transcriptome, but this method cannot fully display the true information of the transcriptome because of its short sequencing read and removal of base modification information. In this study, we used Nanopore direct RNA sequencing technology to isolate and sequence the original poly(A) RNA from the human B lymphocyte line GM12878. A total of 9.9 million poly(A) RNA sequences that passed quality control were generated. The read length N50 was about 1,334bp ( figure 1).
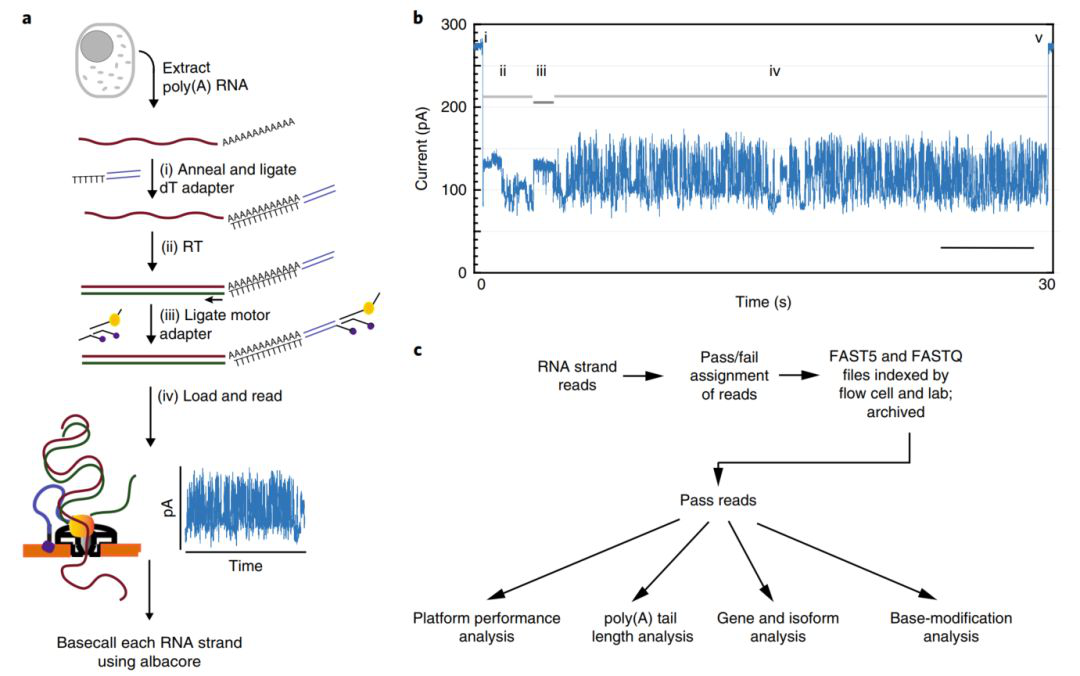
Figure 1 Nanopore natural poly(A) RNA sequencing process
Long nanopore read length can improve the resolution of the exon-exon junction region, thereby mining unannotated RNA isoforms. Using the above long-read data, the researchers first detected and analyzed transcriptional isoforms. Among 33,984 isoforms representing 10,793 genes, 52.6% of unannotated splice junctions were found, and thousands of current splice junctions were identified. Unannotated genes in GENCODE v27, and allele-specific isoforms that cannot be detected using short-read sequencing have been found. For example, for the IFIH1 gene, the paternal isotype retains exon 8 while the maternal isotype does not retain exon 8 (Figure 2).
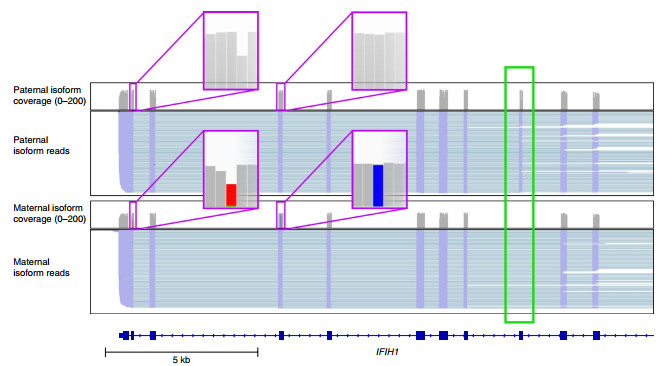
Figure 2 IGV view of ONT direct RNA sequencing transcripts identifying allele-specific isoforms of IFIH1.
The purple box indicates the location of the allele-specific snp (gray is reference, red and blue are SNPs), and the green box indicates alternatively spliced exons.
Then combined with the short-read length data, the research team directly measured the poly(A) tail length, and analyzed the difference between the poly(A) tails of the isomers. Using ONT direct RNA sequencing to estimate the poly(A) tail length of the transcript of the RNA-binding protein DEAD-box helicase 5 (DDX5), the median poly(A) tail length of the intron-retaining isoform is 327nt, while the median poly(A) tail length of its protein-encoding isoform is 125 nt (Figure 3)
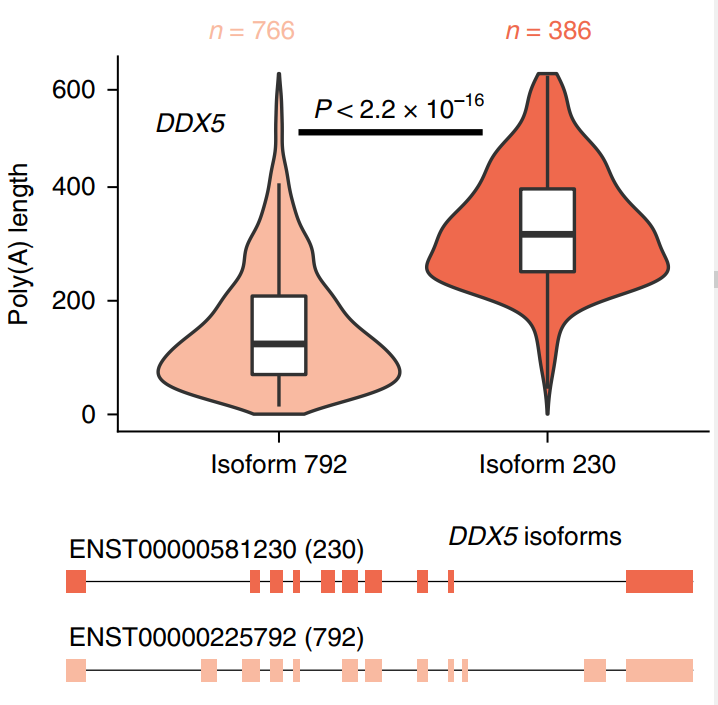
Figure 3 Poly(A) tail length distribution and gene model of two transcript isoforms of DDX5 gene
Finally, using the RNA modification information contained in the direct RNA sequencing data, the researchers screened the changes in the GGACU motif of the GENCODE-sensitive subtypes of ion currents, and found the currents of 86 genes (198 isoforms). The change can be attributed to the isoform-specific m6A modification.
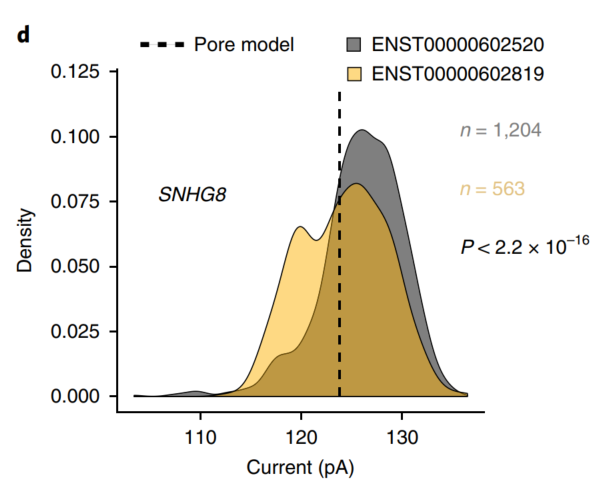
Figure 4 Ion current distribution of GGACU motif in SNHG8 gene isoforms
The researchers showed that RNA modification analysis based on short-read sequencing removes the characteristics between modifications and between modifications and other RNAs, while Nanopore direct RNA sequencing has the ability to detect such remote interactions. References:Workman R E, Tang A D, Tang P S, et al. Nanopore native RNA sequencing of a human poly (A) transcriptome[J]. Nature methods, 2019: 1-9.
 GrandOmics collaborates with Oxford Nanopore to deliver...2019-04-26 - pm2:11
GrandOmics collaborates with Oxford Nanopore to deliver...2019-04-26 - pm2:11 Conference Announcement | January 12-17, GrandOmics sincerely...2024-01-12 - pm3:40
Conference Announcement | January 12-17, GrandOmics sincerely...2024-01-12 - pm3:40 GrandOmics X PacBio Revio – Higher throughput, more...2023-06-09 - pm7:06
GrandOmics X PacBio Revio – Higher throughput, more...2023-06-09 - pm7:06 NextOmics: the FIRST officially certified service provider...2018-01-18 - pm3:00
NextOmics: the FIRST officially certified service provider...2018-01-18 - pm3:00
Follow us on WeChat

GrandOmics

Grandomics Clinical Services

Grandomics Research Services