PJ | 中南林科大、华中农大等联合破译狭叶油茶基因组,为解析油茶自交不亲和、油脂合成等重要性状的形成与调控提供重要参考
2022年3月20日,中南林业科技大学袁德义/张琳油茶科研创新团队,联合华中农业大学金双侠课题组等单位,在经典的国际植物学TOP期刊The Plant Journal上发表了题为“Chromosome-level genome of Camellia lanceoleosa provides a valuable resource for understanding genome evolution and self-incompatibility”的研究论文。希望组在本文章中提供了基因组测序及Hi-C测序服务!
油茶是我国重要的木本食用油料树种,在推进地方经济绿色增长、维护国家食用油安全和乡村振兴中占有十分重要的地位。茶油中不饱和脂肪酸含量达90%以上,还含有丰富的角鲨烯、维生素E、谷甾醇等物质,具有软化血管、降低血脂和血压的作用,是目前国内外最优质的植物食用油。目前,油茶主栽品种主要为多倍体的普通油茶(Camellia oleifera),由于油茶基因组大且亚基因组间的同源异源构成非常复杂,使得多倍体油茶基因组的解析非常困难,严重阻碍了油茶的分子遗传改良。狭叶油茶(Camellia lanceoleosa)是油茶组唯一的二倍体野生种,和多倍体普通油茶亲缘关系最近,破译狭叶油茶基因组不仅可以深入挖掘油茶资源中的优异性状,而且为油茶重要功能基因挖掘利用奠定了坚实基础,使得油茶育种不再是盲人摸象,从而开启了油茶分子育种育种时代。
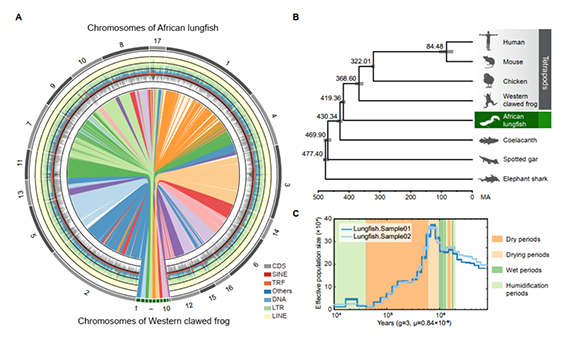
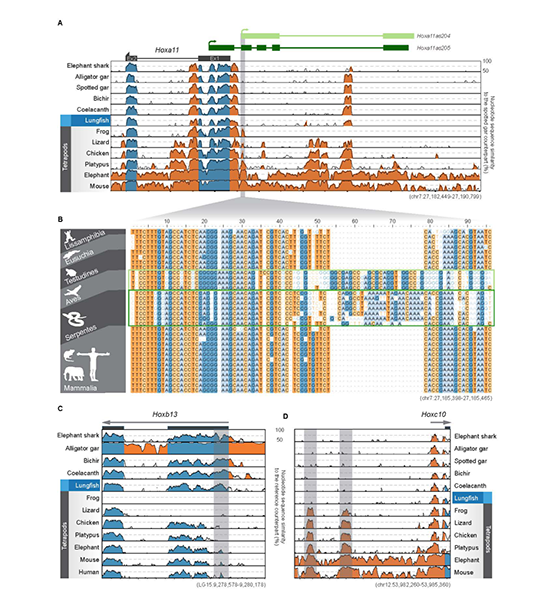
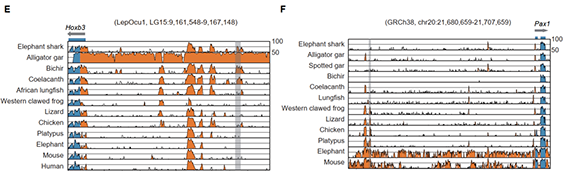
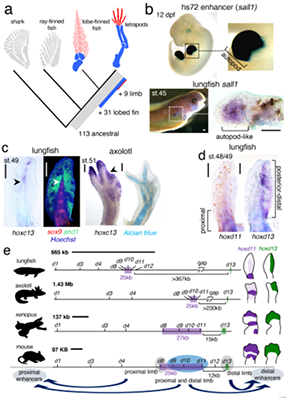
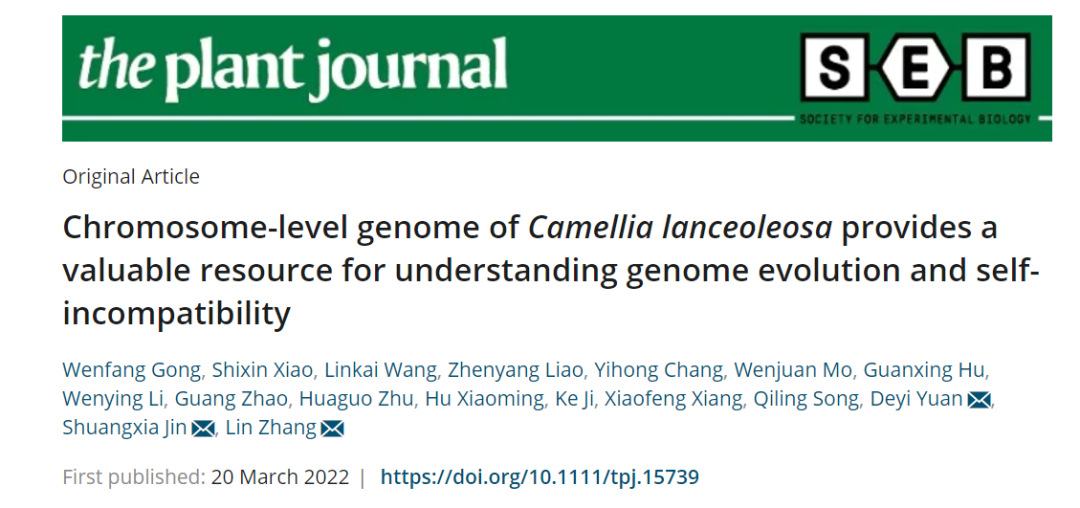
研究团队利用三代Nanopore、二代Illumina和HIC技术,获得了狭叶油茶(2n=30)高质量染色体水平基因组。基因组大小约为3.00 Gb,杂合率高达2.2%,91.85%的序列被挂载到15条染色体上,共注释到54,172个基因。狭叶油茶重复序列占基因组的80.63%,其中转座子占比78.53%。深入分析发现,长末端反转录转座子家族长期而缓慢的扩增及在过去2百万年内的爆发式扩张,加之缺少快速有效的DNA删除机制,最终导致狭叶油茶基因组变得庞大。狭叶油茶与茶叶均为山茶科山茶属二倍体植物。比较基因组研究发现,狭叶油茶与茶叶共享最近的一次WGD事件,并在6-7百万年前发生分化,2号和11号染色体在狭叶油茶与茶叶间存在较大的倒位,这可能是油茶和茶叶在基因组结构上的一个重要变异。
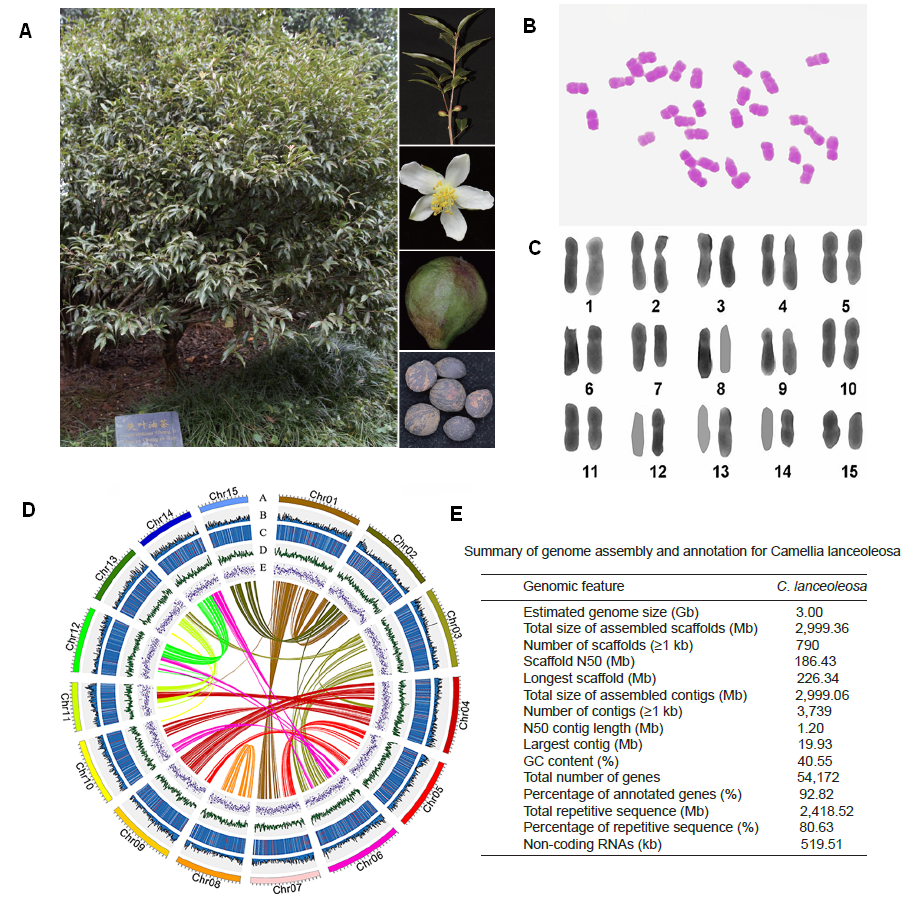
图1 狭叶油茶基因组的组装

图2 狭叶油茶转座子的插入与移除
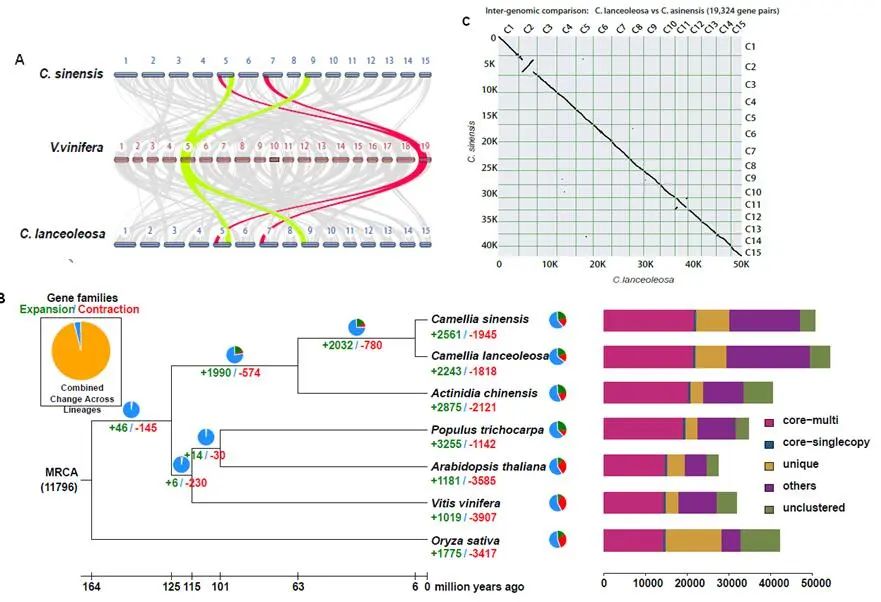
图3 狭叶油茶比较基因组分析
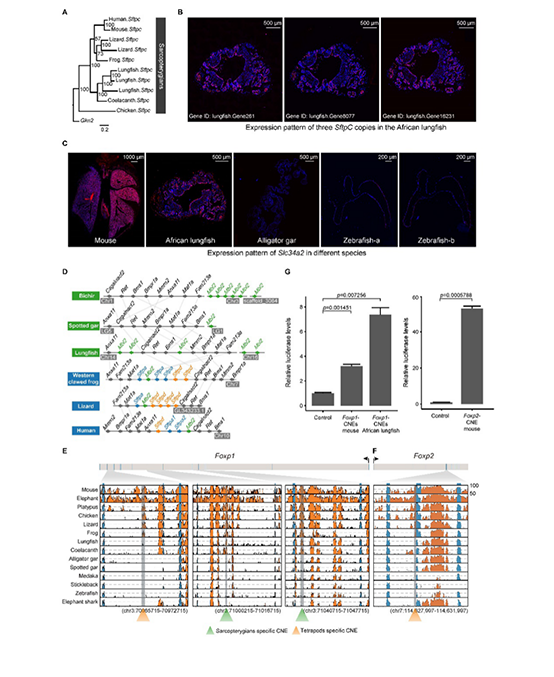
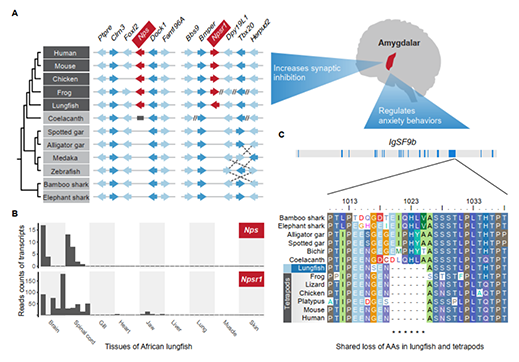
基于狭叶油茶基因组,结合GC_MS以及转录组分析,发现油脂合成的关键基因ACC的扩张及DGAT、GPD、SAD在种子的偏向性表达是狭叶油茶高油脂和高油酸含量的重要原因。儿茶素、茶氨酸、咖啡碱是茶叶品质的重要组成成分,基于UPLC-MS/MS技术的代谢组检测到狭叶油茶叶片也富含儿茶素和茶氨酸,而咖啡碱主要富集在种皮和根中,多组学分析结果表明,SAM-dependent N-methyltransferases与咖啡碱的积累与分布密切相关。细胞学分析显示,狭叶油茶也是后期自交不亲和植物。结合亲和性相关基因的表达、结构特征及染色体定位,解析了狭叶油茶的自交不亲和性特征。狭叶油茶的油脂含量、脂肪酸比例以及次生代谢物分布与含量都与普通油茶类似,以上性状的解析为理解普通油茶油脂合成等重要经济性状的形成与调控提供了重要参考。
图4 狭叶油茶自交不亲和特征解析
中南林业科技大学龚文芳副教授、肖诗鑫讲师及在读硕士生王林凯为论文共同第一作者,中南林业科技大学袁德义教授、张琳教授和华中农业大学金双侠教授为论文共同通讯作者,黄冈师范学院朱华国教授、胡孝明教授及中国农业科学院农业基因组研究所廖振阳博士后也参与了本项研究。本研究得到了国家重点研发计划项目(2018YFD1000603)、湖南省自然科学基金(2020JJ5968)的联合资助。
原文链接:
https://onlinelibrary.wiley.com/doi/epdf/10.1111/tpj.15739










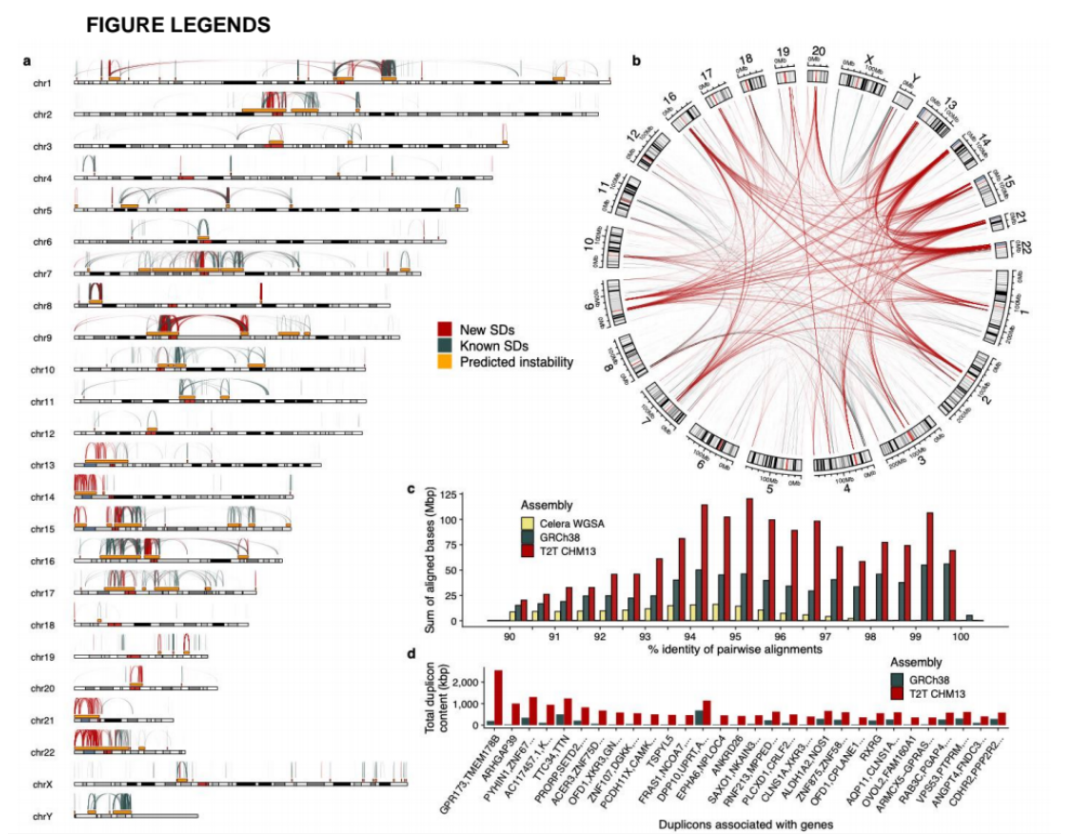



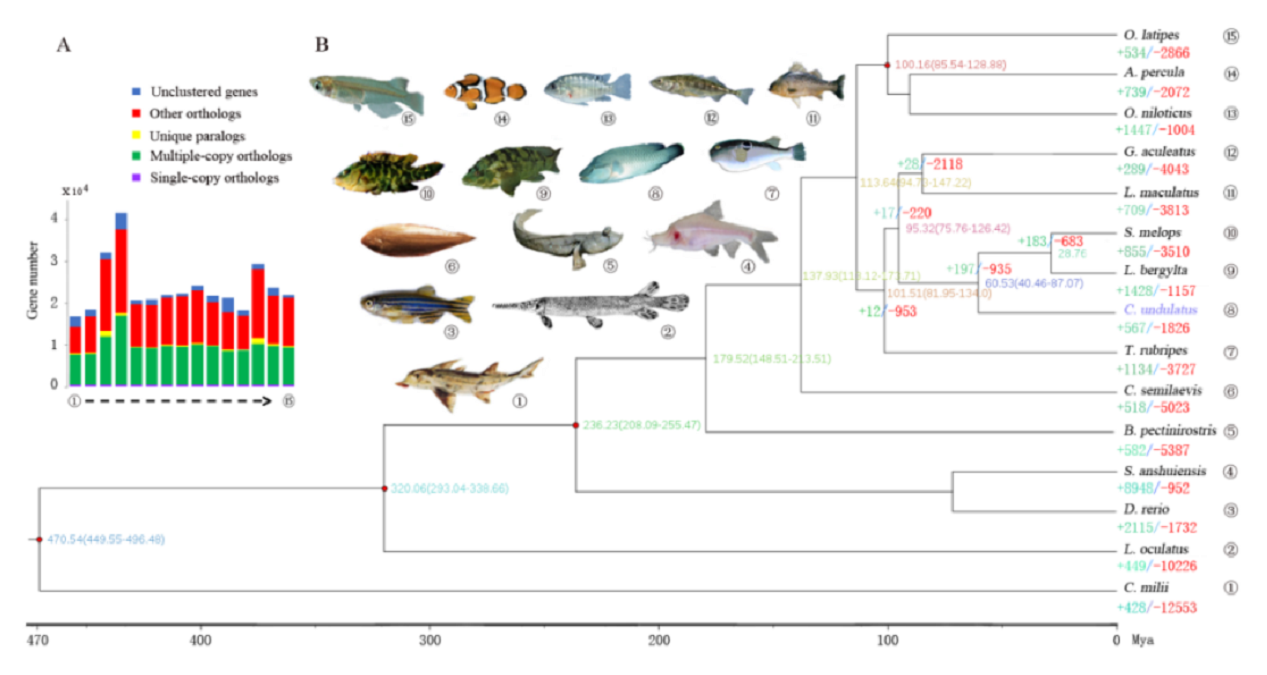
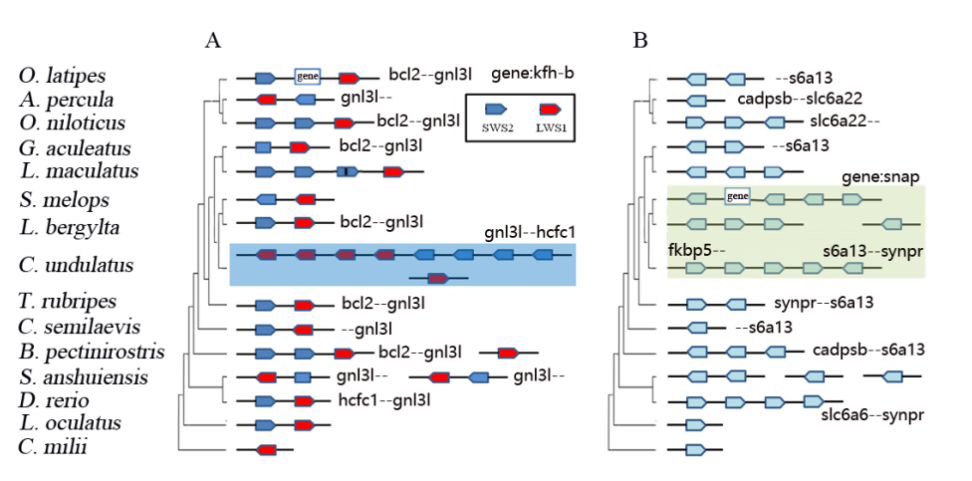
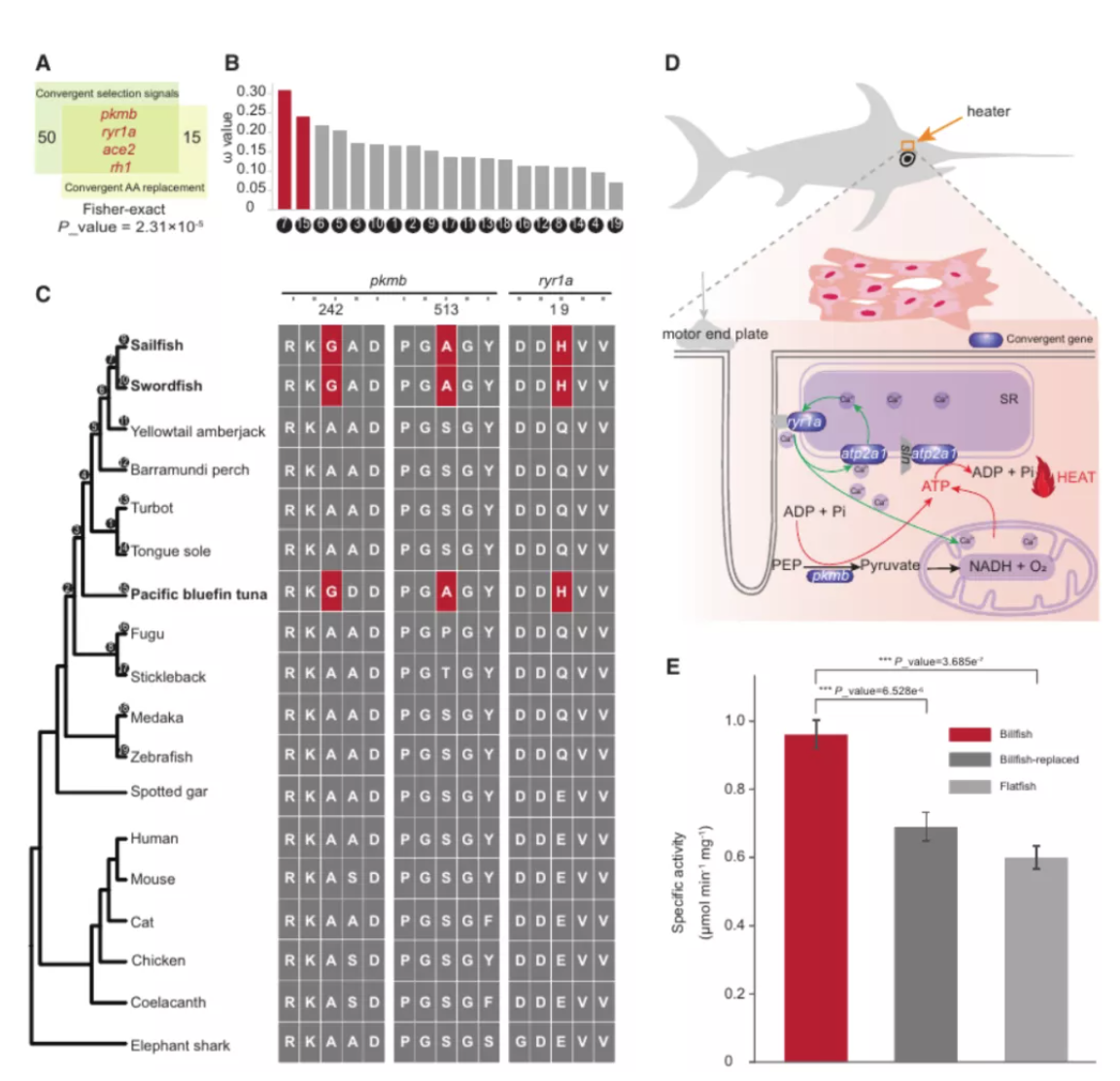

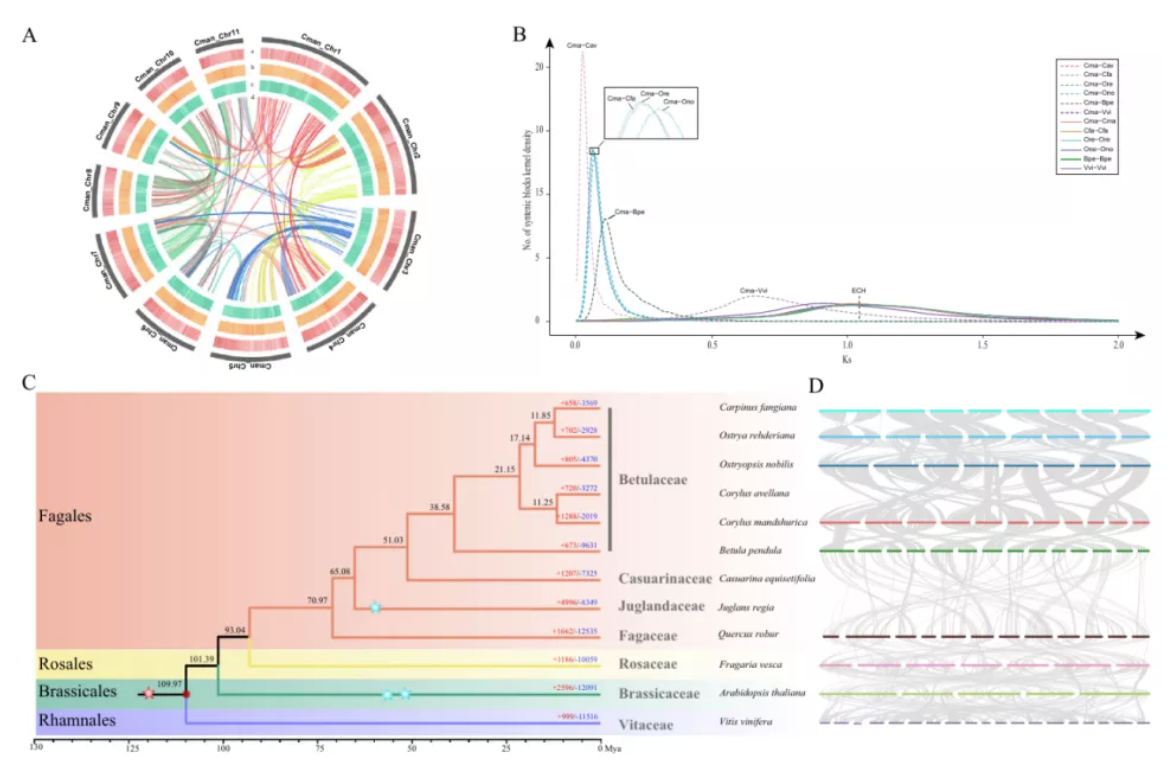
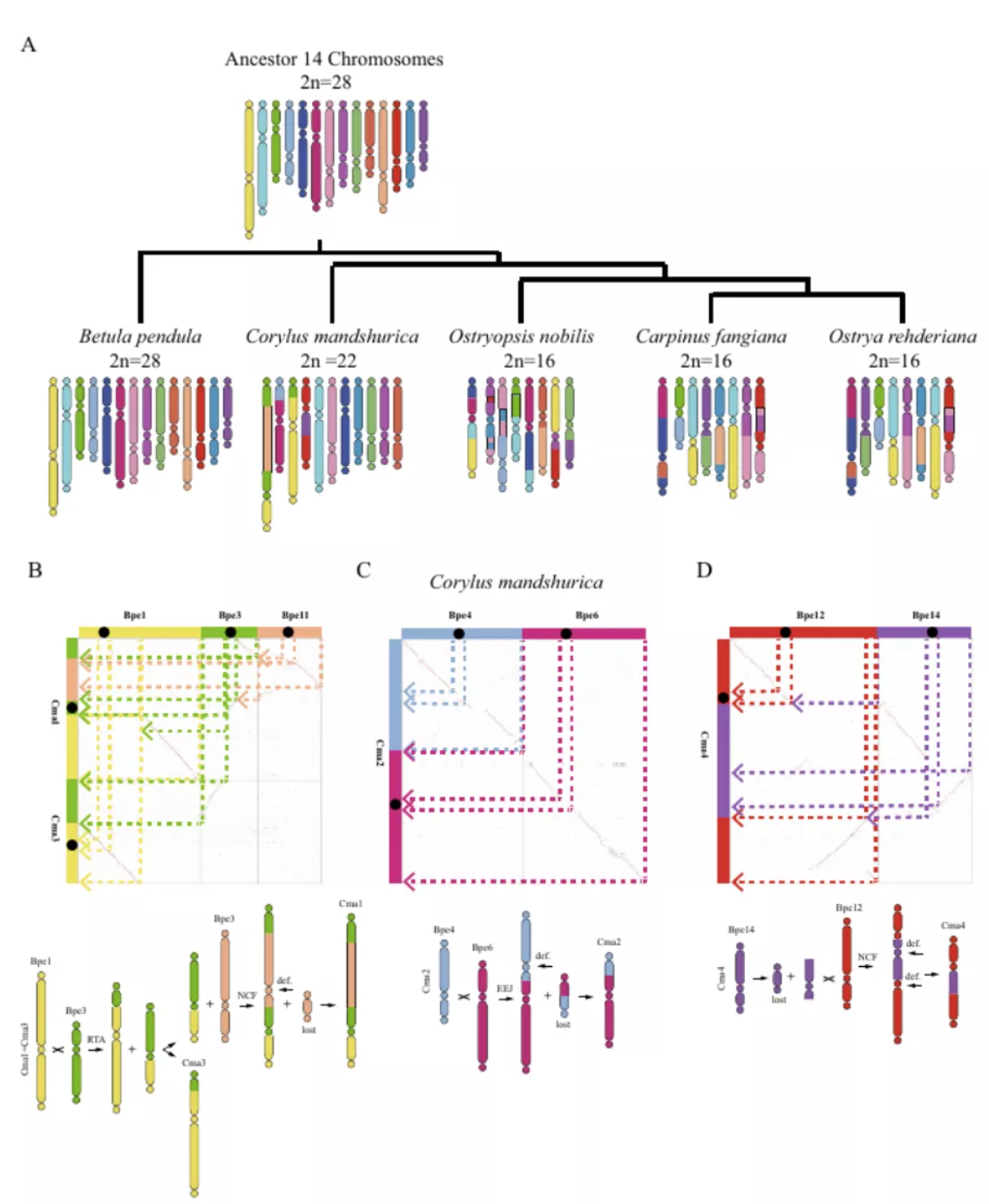
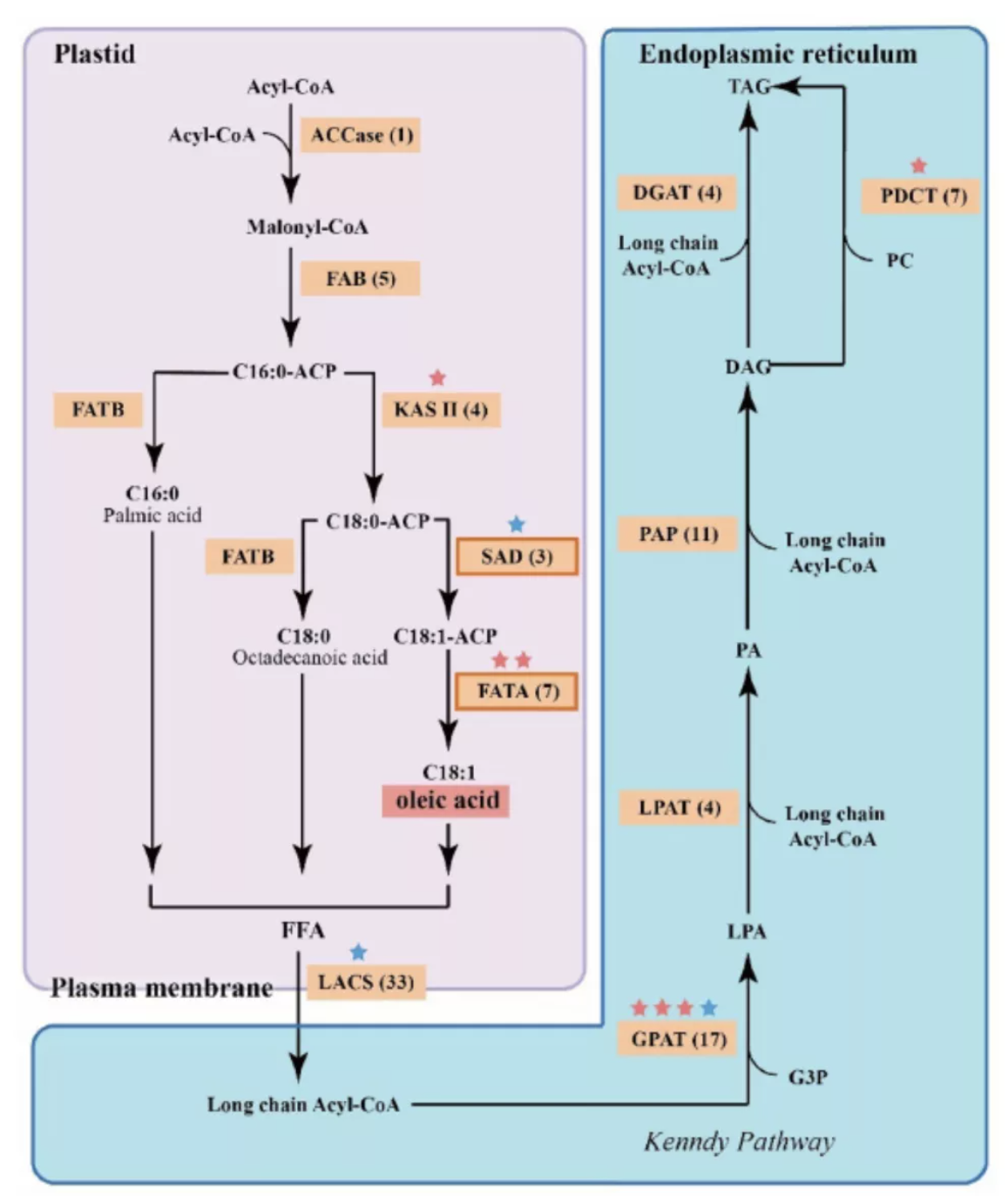

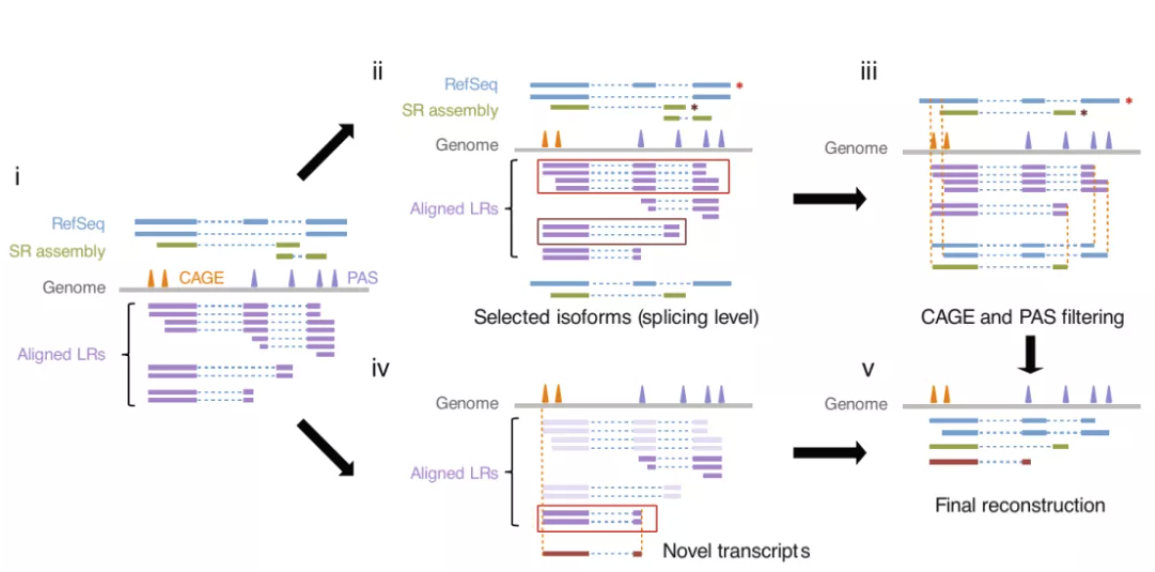
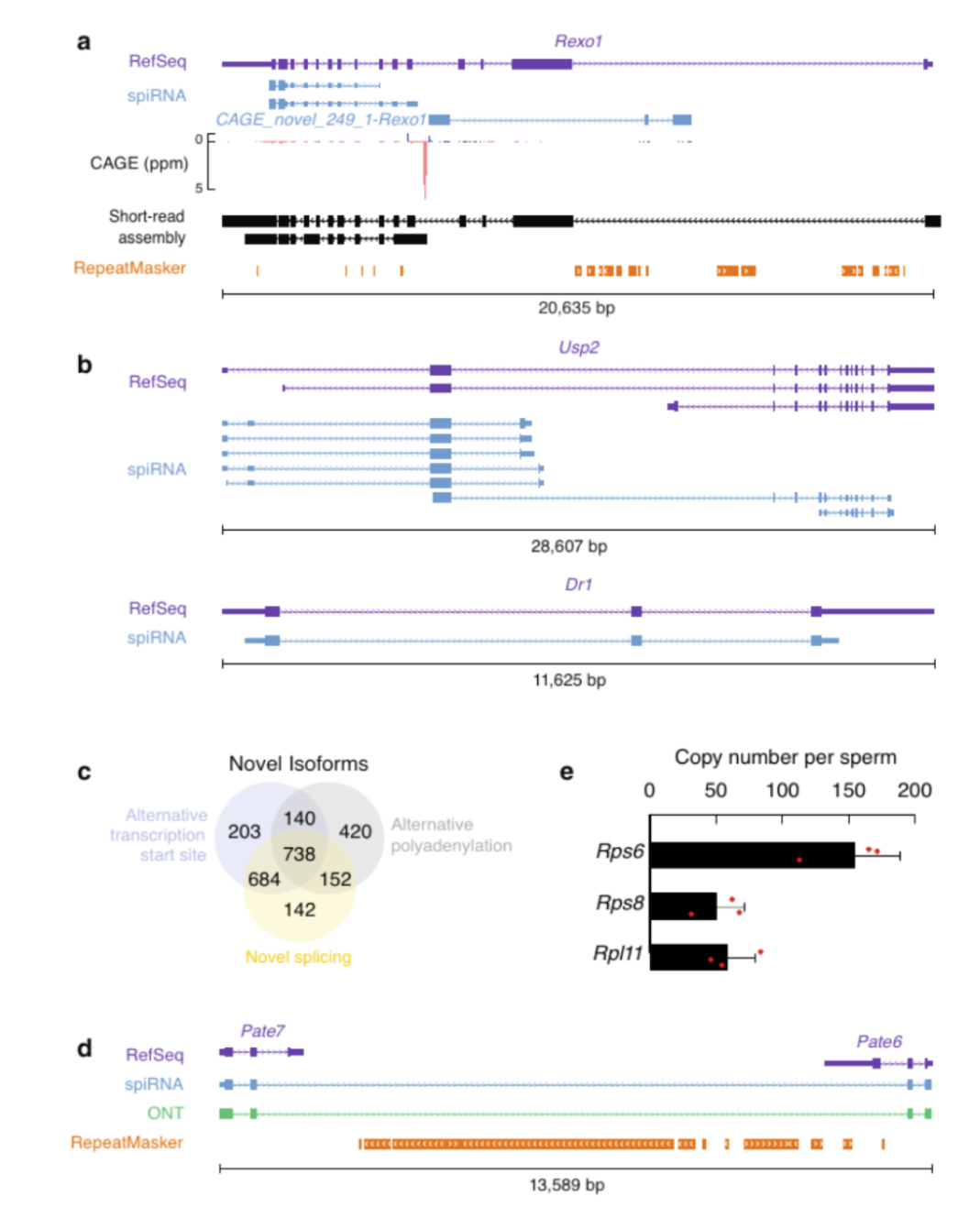
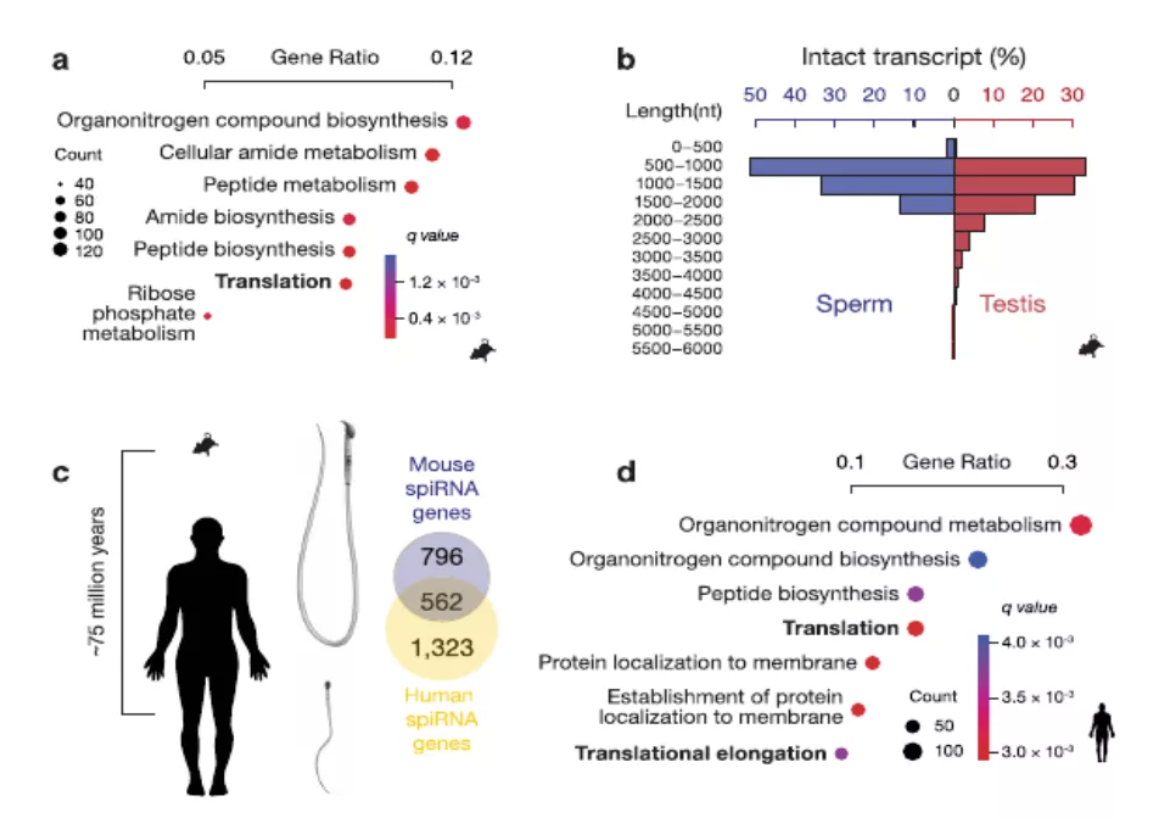
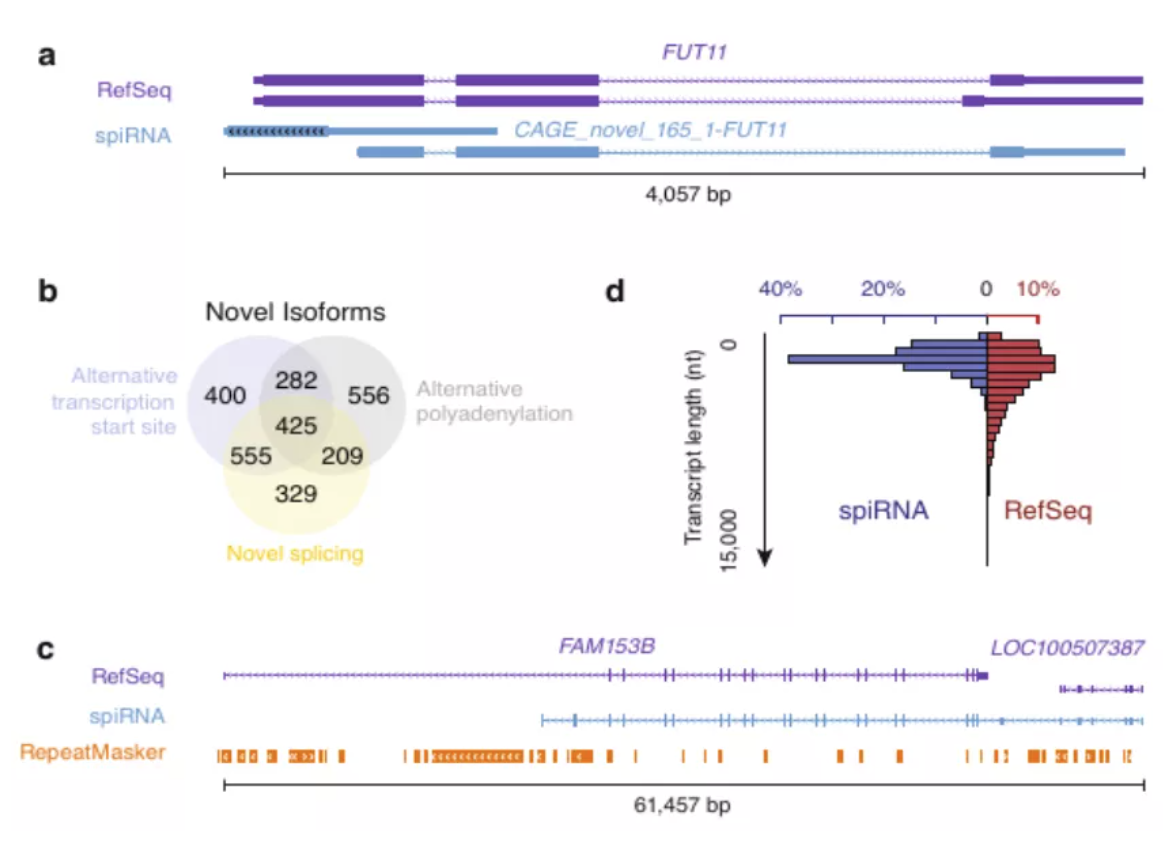
 图3 spiRNAs include both mRNAs and lncRNAs
图3 spiRNAs include both mRNAs and lncRNAs