项目文章 | 首个绞股蓝T2T基因组,解析达玛烷型三萜皂苷生物合成机制
绞股蓝(Gynostemma pentaphyllum),一种葫芦科的多年生藤蔓植物。在医学上有超过600年应用历史,它是超过200种达玛烷型皂苷的宝贵自然来源,具有显著的生物活性,如抗癌、心脏保护、肝脏保护、神经保护和抗糖尿病作用。
研究发现,尽管绞股蓝与人参(Panax ginseng)在进化关系上相距甚远,但其含有人参皂苷和其他结构类似的达玛烷三萜类化合物,同时绞股蓝在获取难度和达玛烷型皂苷含量方面相比人参具有优势。因此,绞股蓝在开发达玛烷型皂苷衍生药物方面,具有极大的应用前景。
2024年4月30日,中国医学科学院药用植物研究所郭宝林/孙超团队在Plant Communications期刊上在线发表了题为“Insights into the dammarane-type triterpenoid spaonin biosynthesis from the telomere-to-telomere genome of Gynostemma pentaphyllum”的研究论文。该论文组装完成了葫芦科绞股蓝的高质量从端粒到端粒(T2T)基因组,初步探究了绞股蓝中达玛烯二醇-II合酶的催化机制,并揭示了绞股蓝和人参中的达玛烯二醇合酶为独立进化而来。希望组为本研究提供了ONT超长、Bionano测序和T2T组装服务。
1.高质量绞股蓝T2T基因组组装
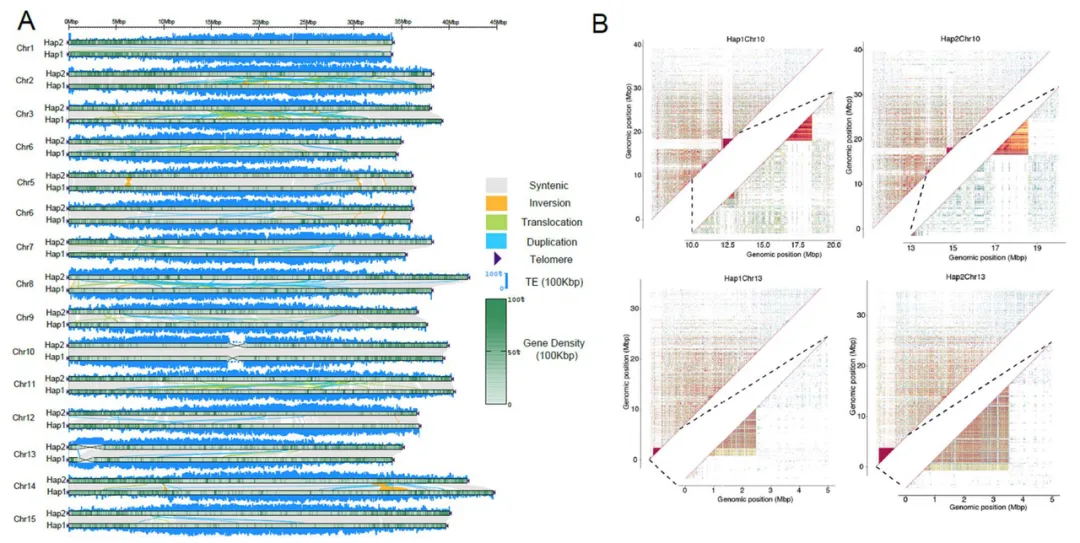
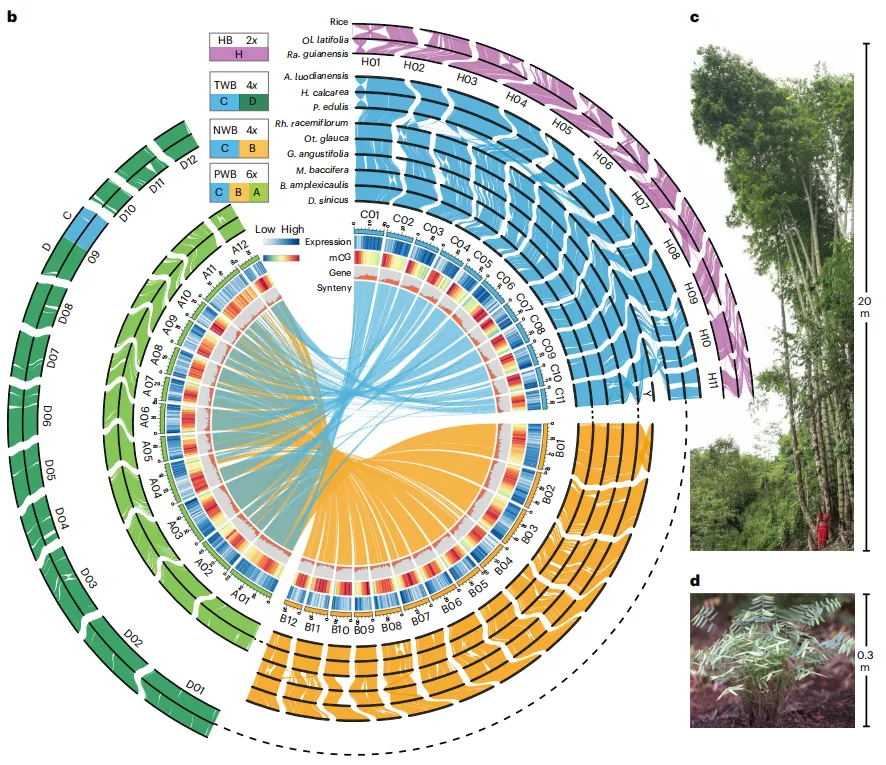
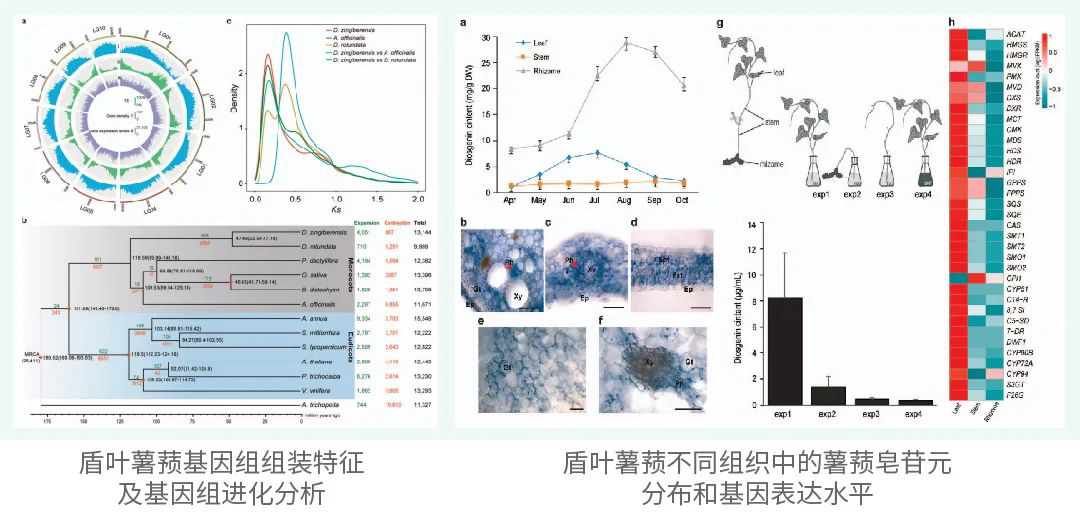
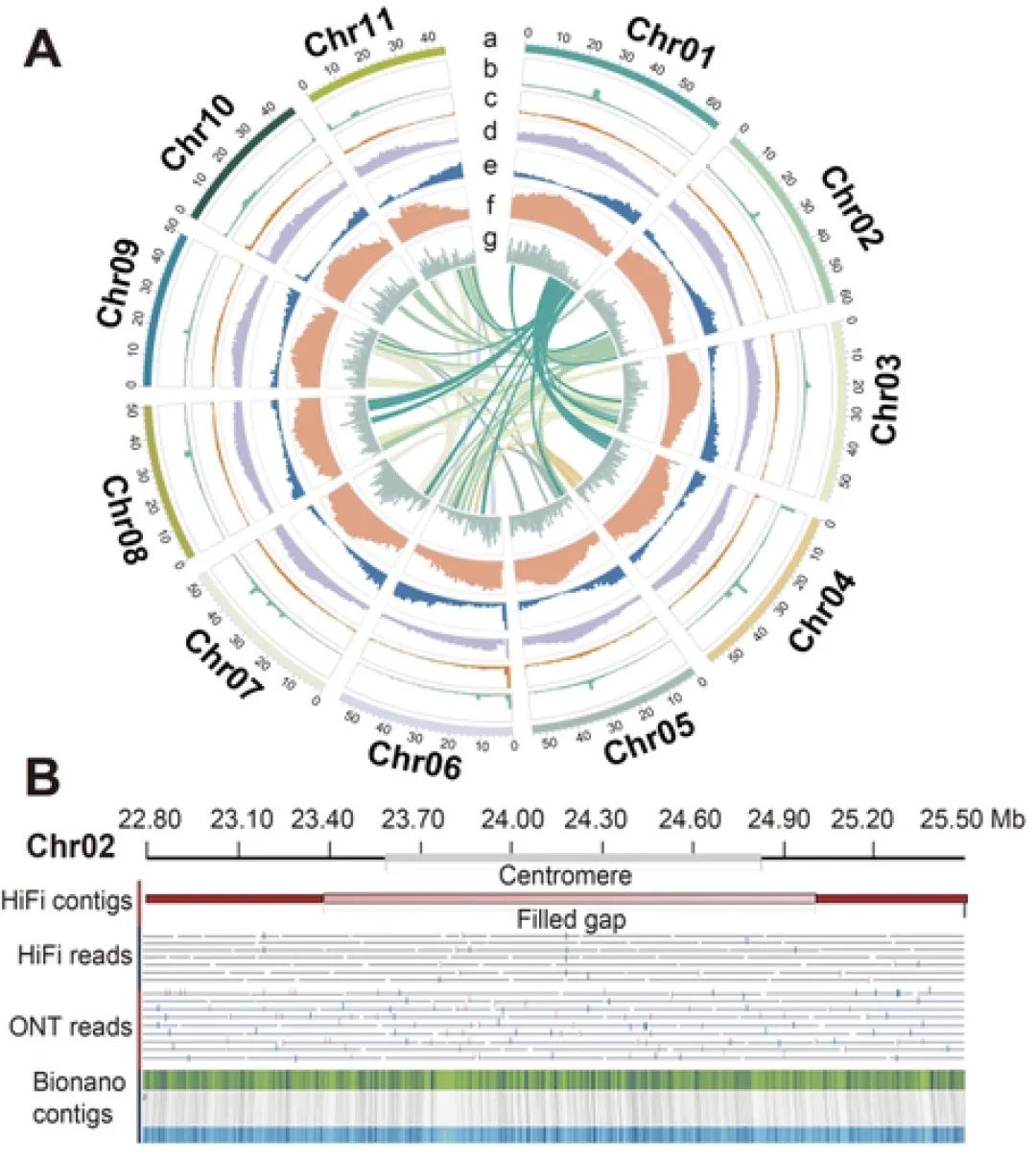
组装使用了30.54 Gb(~51x)的PacBio HiFi数据,103.71 Gb(~173x)的ONT超长数据,65.63 Gb(~109x)的Hi-C以及Bionano数据,最终生成了一个高质量绞股蓝端粒到端粒(T2T)基因组(Gp_T2Tv1.0),其总长度为599.38 Mb,成功识别了11条染色体的所有22个端粒(CAATAAn)和11个着丝粒。
通过多种方法对Gp_T2Tv1.0的准确性和完整性进行评估,BUSCO评估基因组完整性达到98.70%,LAI值为14.89,11条染色体的平均Qv值达到36.57,这些表明了基因组组装碱基水平的准确性和完整性。Illumina、HiFi、ONT超长reads的比对率均超过99%,且Bionano光学图谱在所有组装染色体上均显示出高度一致性。在基因组中预测了26,003个蛋白编码基因,平均编码序列大小为4,567 bp,平均每个基因有5.38个外显子。此外,着丝粒区域主要由68%的转座子元件和32%的串联重复序列组成,这些区域在11条染色体之间的长度和结构组成上存在显著差异。
2.GpOSC1通过环化催化2,3-氧化鲨烯形成达玛烯二醇-II的机制
在萜类化合物生物合成中,由氧化鲨烯环化酶(OSCs)催化的2,3-氧化鲨烯的环化是萜类化合物合成的第一个关键的分支点。在对绞股蓝的T2T基因组分析中,一共注释了十一个OSCs。通过酵母表达系统、本氏烟草的瞬时基因表达系统以及相色谱-质谱(GC-MS)分析证明,GpOSC1能够通过环化催化2,3-氧化鲨烯形成达玛烯二醇-II(dammarenediol-II)。
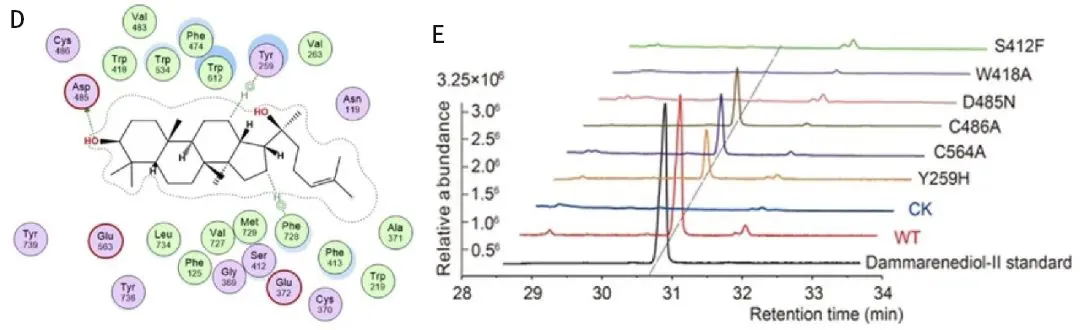
为了更深入地了解GpOSC1(下文称为GpDS)的催化机制,基于GpOSC1的3D结构与达玛烯二醇-II的分子对接结果和保守序列(图1D),构建了GpDS的氨基酸残基Y259H、W418A、D485N、C564A、S412F、H479N和C486A突变体。单点突变体D485N、S412F、W418A导致活性完全丧失,而C486A、C564A、H479N、Y259H突变体的活性显著降低(图1E)。结果表明,GpDS活性位点的几个残基在酶活性中起着至关重要的作用,可能是通过与底物相互作用和塑造整体构象来实现的。这些发现与之前关于人参(P. ginseng)中的达玛烯二醇-II合成酶(DS,ID: ACZ71036.1)的报道相一致,表明GpDS与PgDS具有相似的催化机制。总的来说,我们推测酸性残基D485通过作为质子供体来启动2,3-氧化鲨烯的环化,而C486和C564通过与D485形成氢键来增加其酸性。在VWCYFR motif中的Y259残基对于稳定中间阳离子和促进达玛烯二醇-II的形成至关重要。
3.探寻达玛烯二醇-II合成酶(DS)的起源和进化轨迹
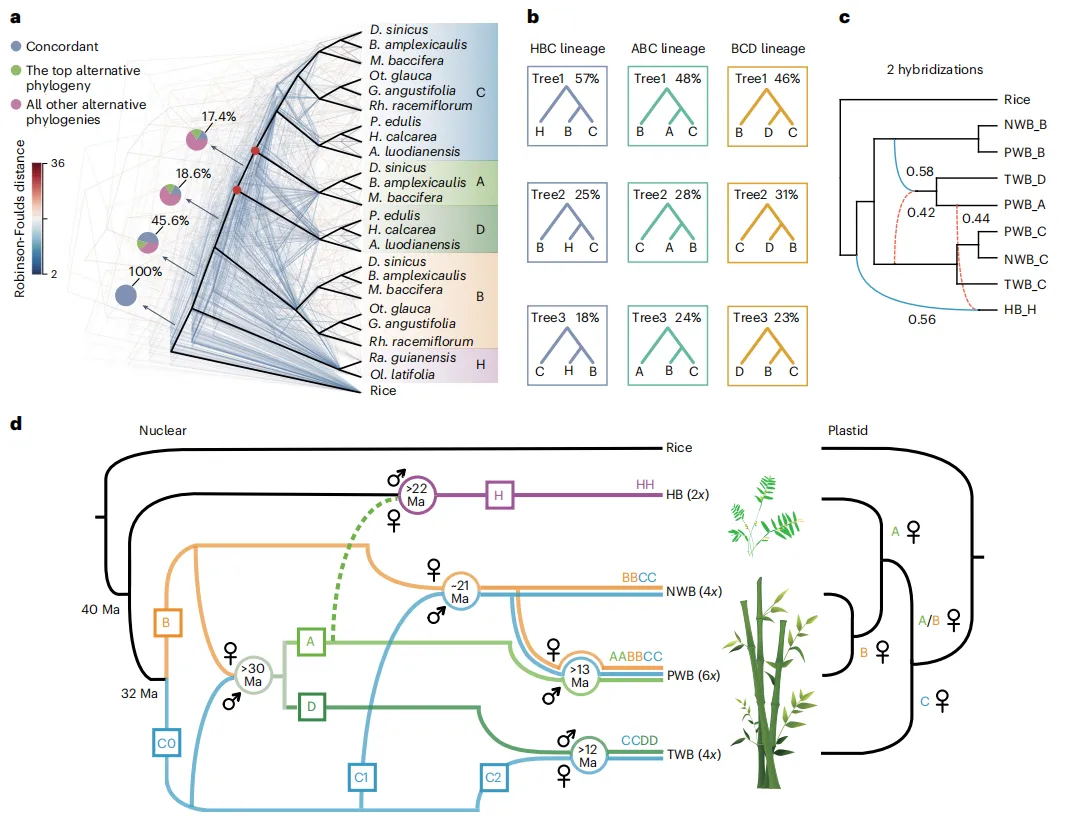
为了研究开花植物中DS的起源和进化轨迹,结合系统发育和共线性分析提出了一个OSC进化的模型(图1F)。通过对来自115个植物的428个OSC序列进行全面的系统发育分析,发现基础被子植物无油樟(Amborella trichopoda)含有一个单独的OSC,它与在蕨类和裸子植物中发现的环阿屯醇合成酶(CASs)具有同源性,表明被子植物中的所有OSC都是从祖先CAS-like蛋白进化而来的。剩余的OSC最初被分为两个主要分支,分别命名为分支A和分支B。每个分支都包含了来自被子植物主要分类群的物种衍生的OSC,这表明大多数被子植物在A. trichopoda分化后共享了一个共同的OSC基因复制事件。复制事件之后,在核心真双子叶植物中,同源OSC基因A和B呈现出了三分支的进化模式,亚分支A1、A2和A3从分支A进化而来,而亚分支B1、B2和B3则从分支B进化而来。OSCs的系统发育分析表明,B2亚分支经历了显著的新功能化,其中葫芦科家族的GpDS位于该亚分支的β-香树脂合成酶(bAS)基因内。相反,在B3亚分支中,来自五加科的PgDS的多功能OSC聚类在一起。因此推测在G. pentaphyllum和P. ginseng中,DS是独立进化的。 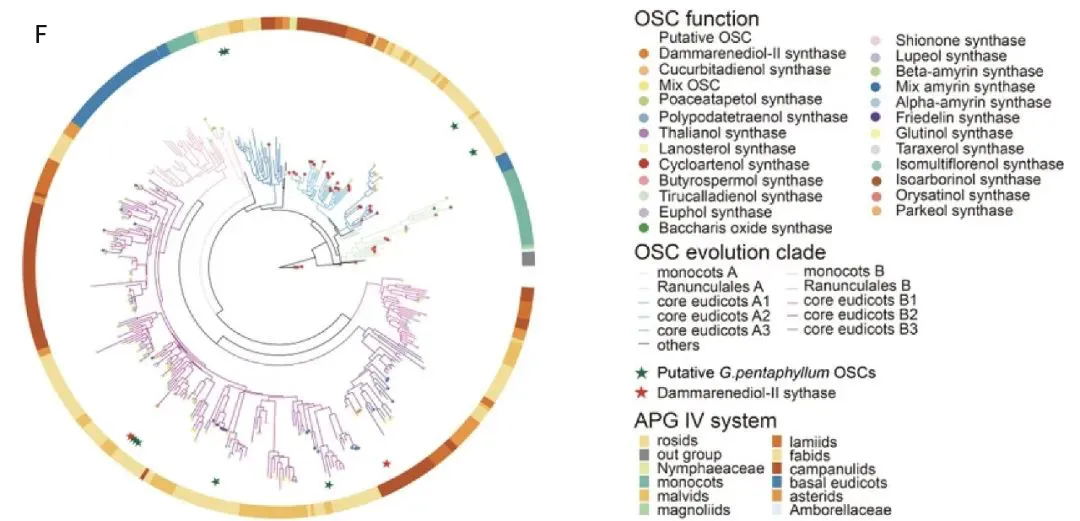
总之,该研究完成了绞股蓝的高质量T2T基因组组装,并对基因组中鉴定出的11个候选OSCs基因中的一个DS进行了功能表征,为解析达玛烷型三萜皂苷生物合成机制提供了参考意义,有利于绞股蓝在达玛烷型皂苷衍生药物方面的开发应用。