组∙而论道 第十五期 | 北大汤富酬教授团队最新技术scNanoHi-C2,文章一作卢健森博士亲自授课!
本期讲座卢健森博士将分享单分子测序平台的单细胞三维基因组构象捕获技术(scNanoHi-C2)及其在早期胚胎生殖细胞发育过程中的应用,为感兴趣的听众提供单细胞水平研究高阶三维基因组结构的参考思路。


本期讲座卢健森博士将分享单分子测序平台的单细胞三维基因组构象捕获技术(scNanoHi-C2)及其在早期胚胎生殖细胞发育过程中的应用,为感兴趣的听众提供单细胞水平研究高阶三维基因组结构的参考思路。
武汉希望组医学检验实验室有限公司 | |
| 组织机构代码 | 91420100MA4KXQ2J1P |
| 法定代表人 | 汪德鹏 |
| 生产地址 | 武汉市东湖新技术开发区花城大道8号武汉软件新城C11栋17楼 |
| 生产经营内容 | (共1个一级诊疗科目)医学检验科(临床免疫、血清学专业,临床细胞分子遗传学专业) |
| 危险废弃物类别 | HW01医疗废物,HW49其他废物 |
| 危废名称 | 医疗废物,废液,废试剂瓶 |
| 危废代码 | 841-001-01,900-047-49 |
| 处置方式 | Y10医疗废物焚烧, |
| 主要成分 | 废弃离心管、废吸头、废试剂瓶、废液和废采血管,一次性医用口罩、手套、帽子等实验用品 |
| 产废工序 | 一次性医用口罩、手套、帽子等实验用品为检验人员使用后医疗废物,废弃离心管、废吸头、废试剂瓶和废采血管为接触过样本或检验试剂的废弃医疗废物。 |
| 危险特性 | 潜在感染性、毒性 |
| 安全措施 | 我机构产生的医疗废物统一使用专用医疗垃圾袋密封,经次氯酸钠喷洒、高温高压等消毒措施处理后集中存放于医疗废物暂存间医疗垃圾桶。医废暂存间由专人进行管理,暂存环境使用次氯酸钠和紫外灯照射消毒。我机构医疗废物委托给武汉汉氏环保工程有限公司处置,废液和废试剂瓶委托华新环境工程(武穴)有限公司处理。 |
| 贮存区域 | 医疗废物暂存间,危废暂存间 |
| 处置去向 | 武汉汉氏环保工程有限公司,华新环境工程(武穴)有限公司 |
| 医废暂存间管理员 | 王飞18171678655 |
| 紧急联系人 | 许力晏18271468344 |
| 相关联系人 | 陶庆15572523735 |
| 产生量(2024年)(单位:吨) | 11.408 |
| 转运量(2024年)(单位:吨) | 11.408 |
| 库存量(2024年)(单位:吨) | 1.684 |

作为专精特新”小巨人”企业,希望组始终深耕单分子测序技术创新,构建起覆盖基因测序方案研发、生信算法开发及医学应用转化的全链条技术体系。此次认定既是对我们自主研发能力的认可,更为企业技术攻关注入了新动能。
希望组自引进PacBio SPRQ化学技术以来,通过近期实验条件优化,并进行了多批次的SPRQ试剂的样本实测,单张芯片产出突破150G!文库长度达到20Kb!接下来让我们看看近期SPRQ试剂带来哪些测序表现吧。

表1 单样本下机数据产出表现
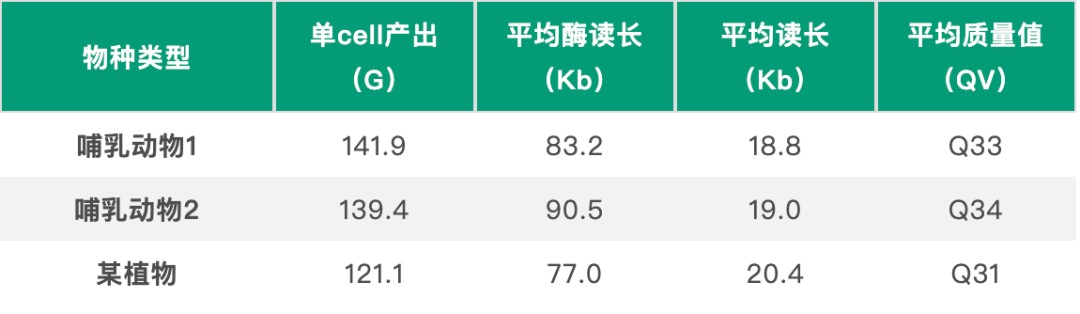
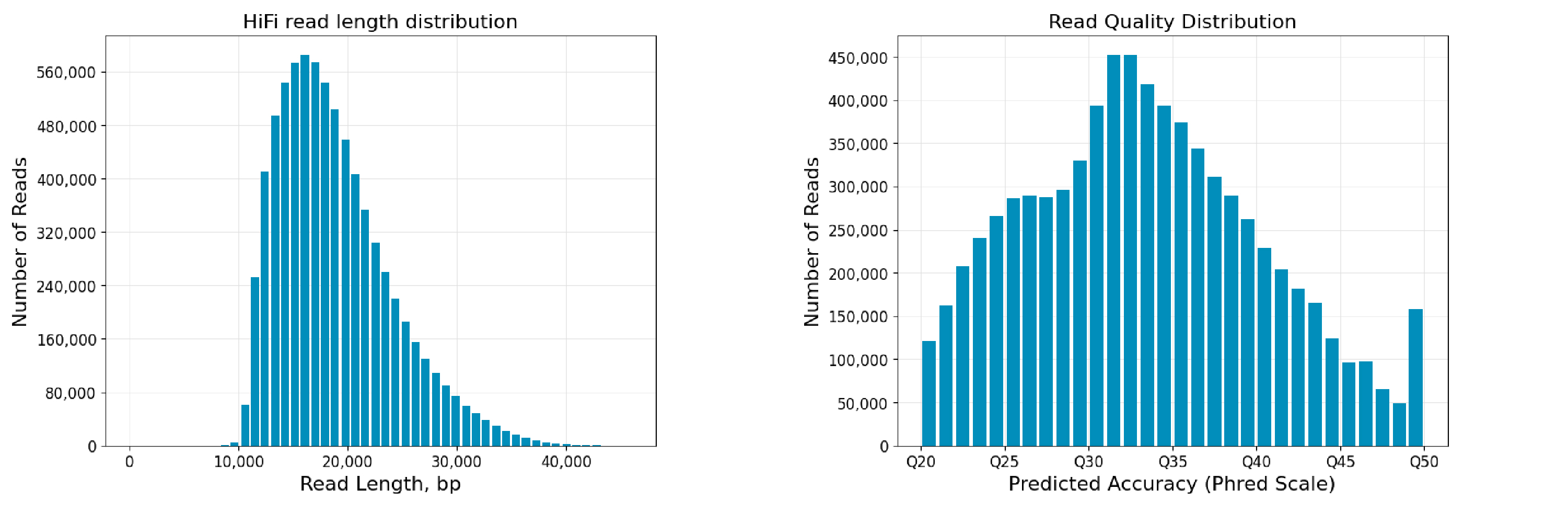
图1 哺乳动物1样本reads长度及质量分布图

图2 哺乳动物2样本reads长度及质量分布图
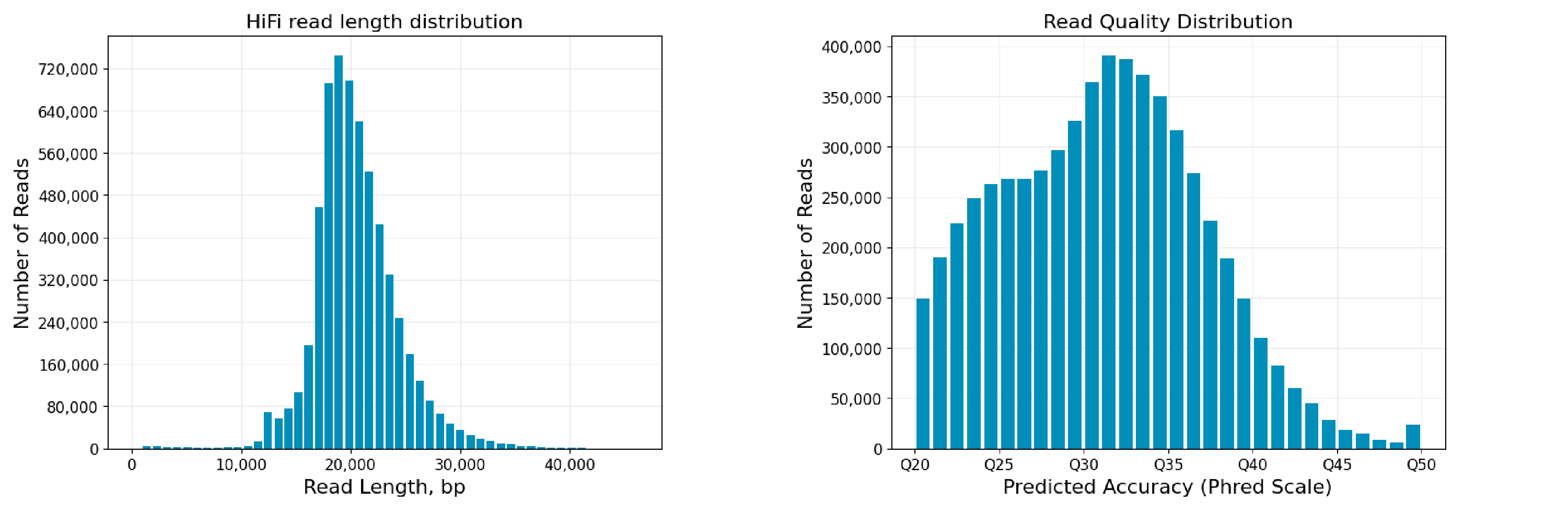
图3 某植物样本reads长度及质量分布图
表2 混样下机数据产出表现
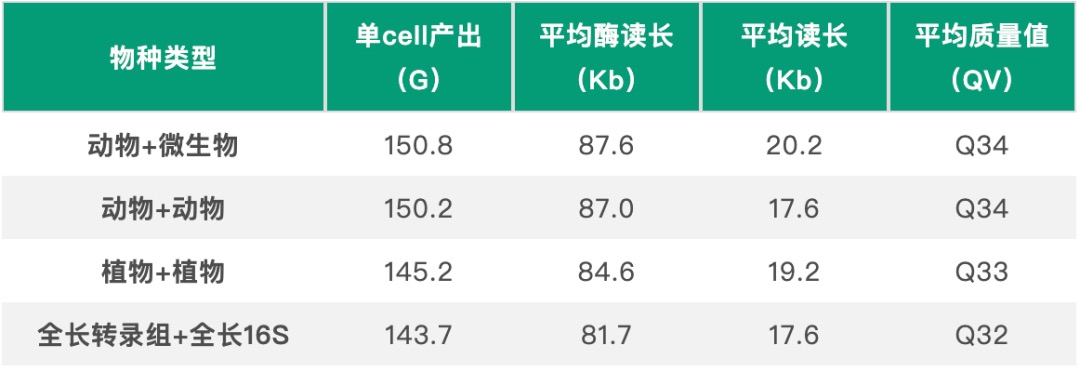
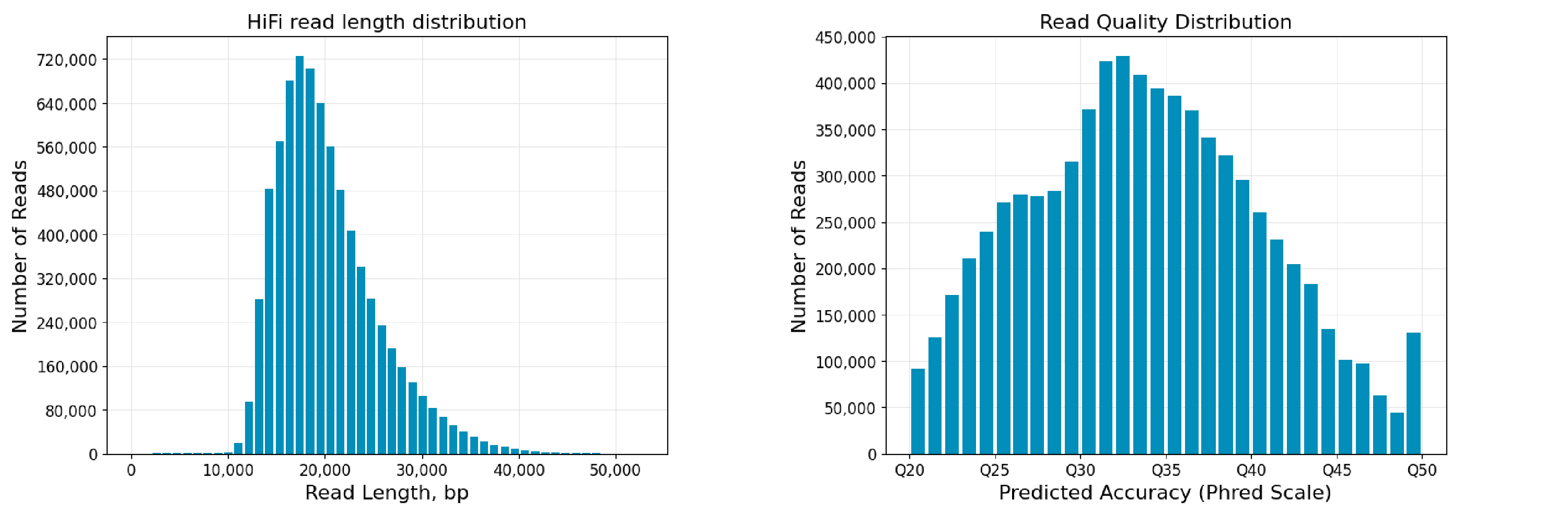
图4 动物+微生物样本混测reads长度及质量分布图
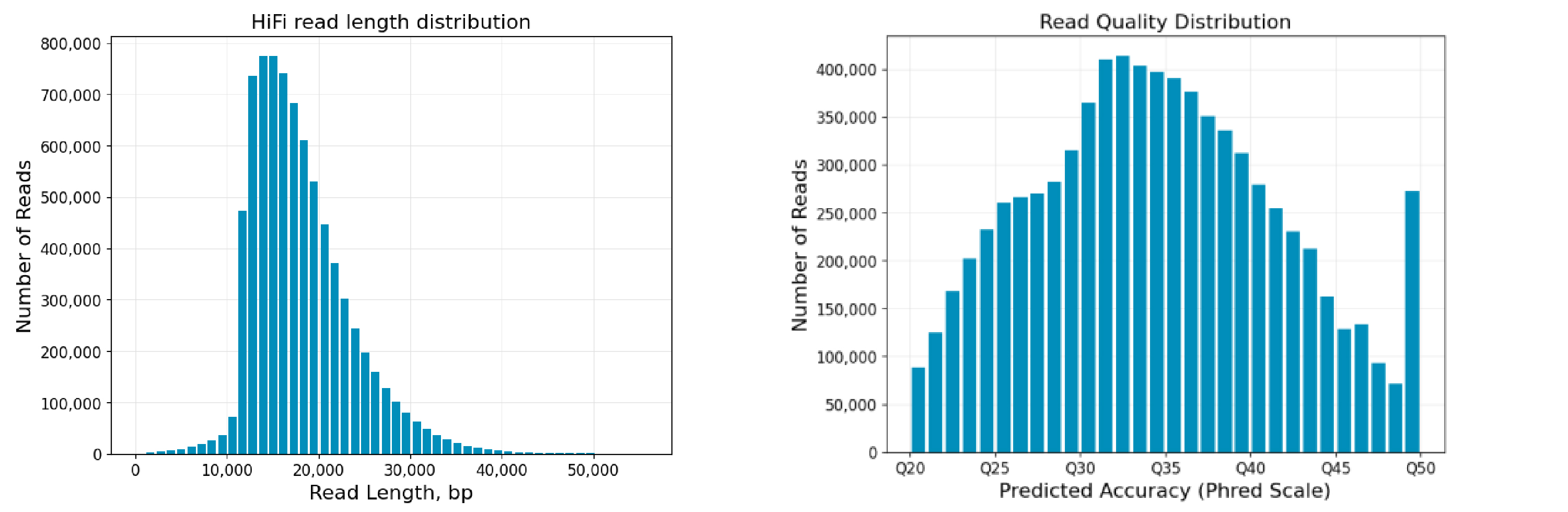
图5 动物+动物样本混测reads长度及质量分布图
图6 植物+植物样本混测reads长度及质量分布图

图7 全长转录组+全长16S样本混测reads长度及质量分布图
Revio平台SPRQ试剂测序结果表现突出,单张cell平均产出高达126.3G,最高产出达到150.8G,reads平均读长达到20Kb。其中,混测样本测序产出表现优异,这为小基因组样本提供极大便利及优惠空间。
希望组基于SPRQ化学技术的强大功能,加上不断进行优化建库测序,致力于为客户提供更卓越的长度长测序服务!




过去的一周中, Oxford Nanopore London Calling顺利召开,会上分享了Oxford Nanopore 产品的最新情况,引入基于Transformer的大语言模型训练了新的basecaller – V5.0.0 SUP,实现了单链读取准确度大幅提升!
ONT官方数据展示
常规马达蛋白E8.2 + SUP basecalling 模型V4.2.0,ONT下机数据碱基质量峰值可达Q22(下文用Q20指代)。
官方升级更新后:新马达蛋白E8.2.1 + SUP basecalling 新模型V5.0.0,实现ONT下机数据碱基质量峰值达到Q28(下文用Q28指代)。
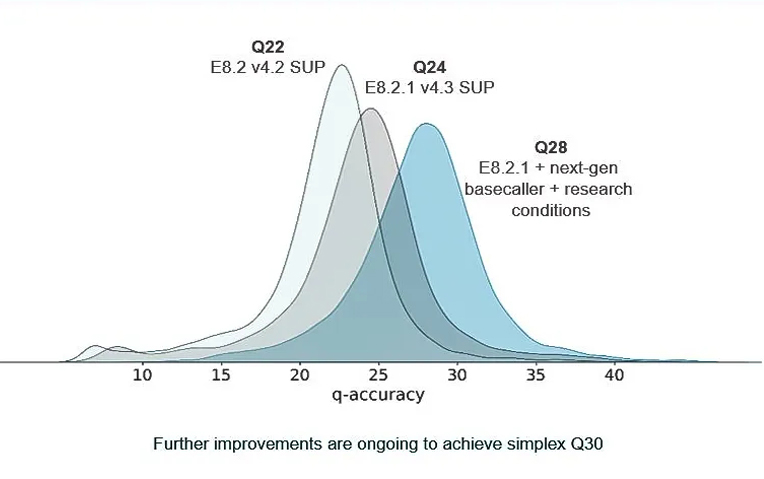
希望组实测数据首发展示
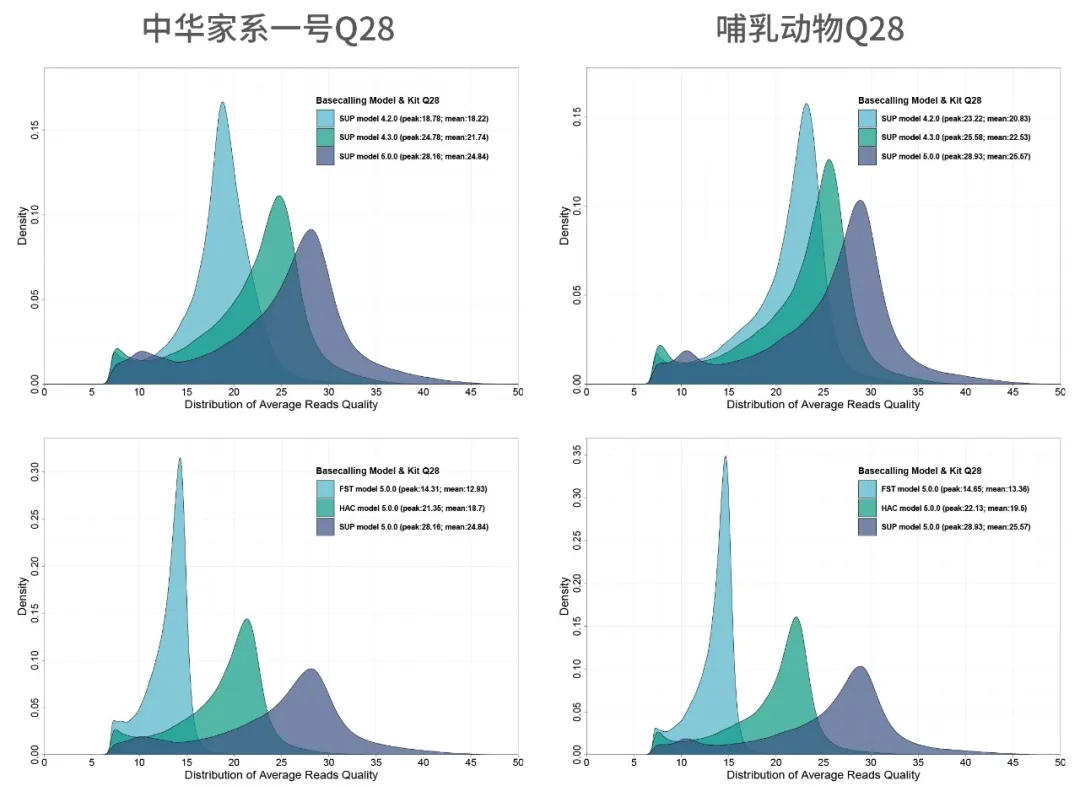
以Q7为过滤标准:Q28马达蛋白+不同basecalling模型比较
Q28马达蛋白+SUP basecalling V5.0.0模型,呈现最优碱基质量均值 & peak值,迈向ONT数据交付新高度
2.Q28马达蛋白+SUP V5.0.0模型,Q10 pass reads高达96%
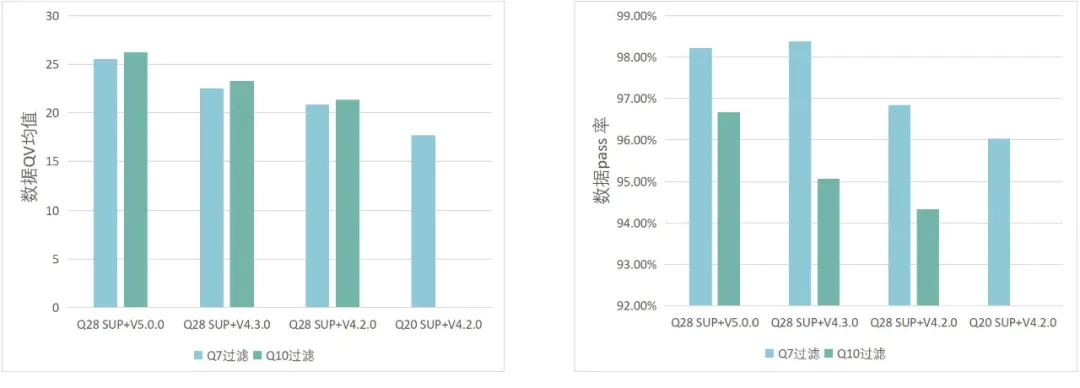
哺乳动物示例:不同过滤标准+不同SUP basecalling模型比较
高质量 & 高产出
Q28马达蛋白+ SUP V5.0.0模型,以Q7为过滤标准,碱基质量均值25+,pass率高达98%
Q28马达蛋白+ SUP V5.0.0模型,以Q10为过滤标准, 碱基质量均值26+,pass率高达96%
即日起希望组承诺:
以Q10为过滤标准,Q28马达蛋白 + SUP V5.0.0
人血液质检合格样本 ONT Ultra-long N50 100K交付数据量不低于20G/Cell!
3. Q28马达蛋白+SUP V5.0.0模型,Q20 reads占比高达80%
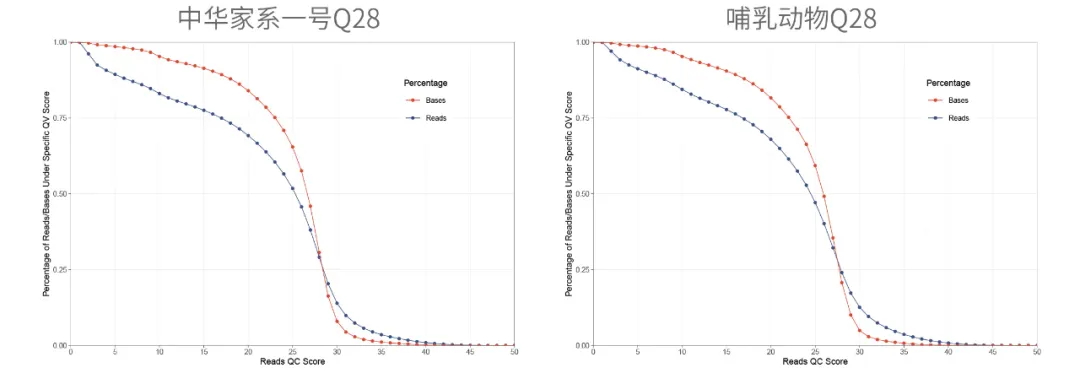
Q28马达蛋白+SUP basecalling V5.0.0模型不同Q值过滤标准下数据占比
即日起希望组承诺:
以Q20为过滤标准,Q28马达蛋白 + SUP V5.0.0
人质检合格样本 ONT Ultra-long N50 100K交付数据量不低于15G/Cell!
16S 扩增子测序:通过对特定环境中的微生物(细菌)DNA特定区域进行扩增测序,以研究微生物群落组成、物种丰度以及样本间群落组成差异情况。
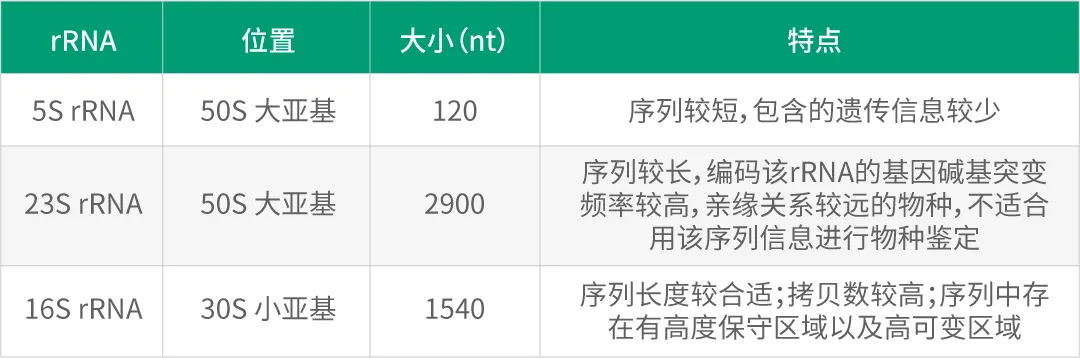
细菌核糖体RNA(rRNA)按沉降系数分为3种,分别为5S、16S和23S rRNA。16S rRNA是细菌核糖体中30S亚基的组成部分,与5S rRNA、23S rRNA相比,其序列长度合适、拷贝数较高以及序列中存在有高度保守区域以及高可变区域。因而,16S rDNA 是目前最适于细菌系统发育和分类鉴定的指标。
二代扩增子测序因测序读长的限制,无法对原核生物16S中所有可变区进行测序,因此一般只能针对1-2个可变区进行测序分析和物种鉴定。部分高可变区所携带的变异信息有限,无法实现种水平的物种注释,导致二代微生物多样性通常在属及属以上的水平进行研究。而三代扩增子主要基于PacBio Revio测序平台,利用单分子实时测序(SMRT)的方法,基于HiFi模式对全长16S(V1-V9)进行测序,获取更多的高可变区信息以了解样本中的物种组成以及相对丰度等,显著提高物种注释的分辨率和准确性,更全面的反映微生物的群落结构。
全长16S扩增子的产品优势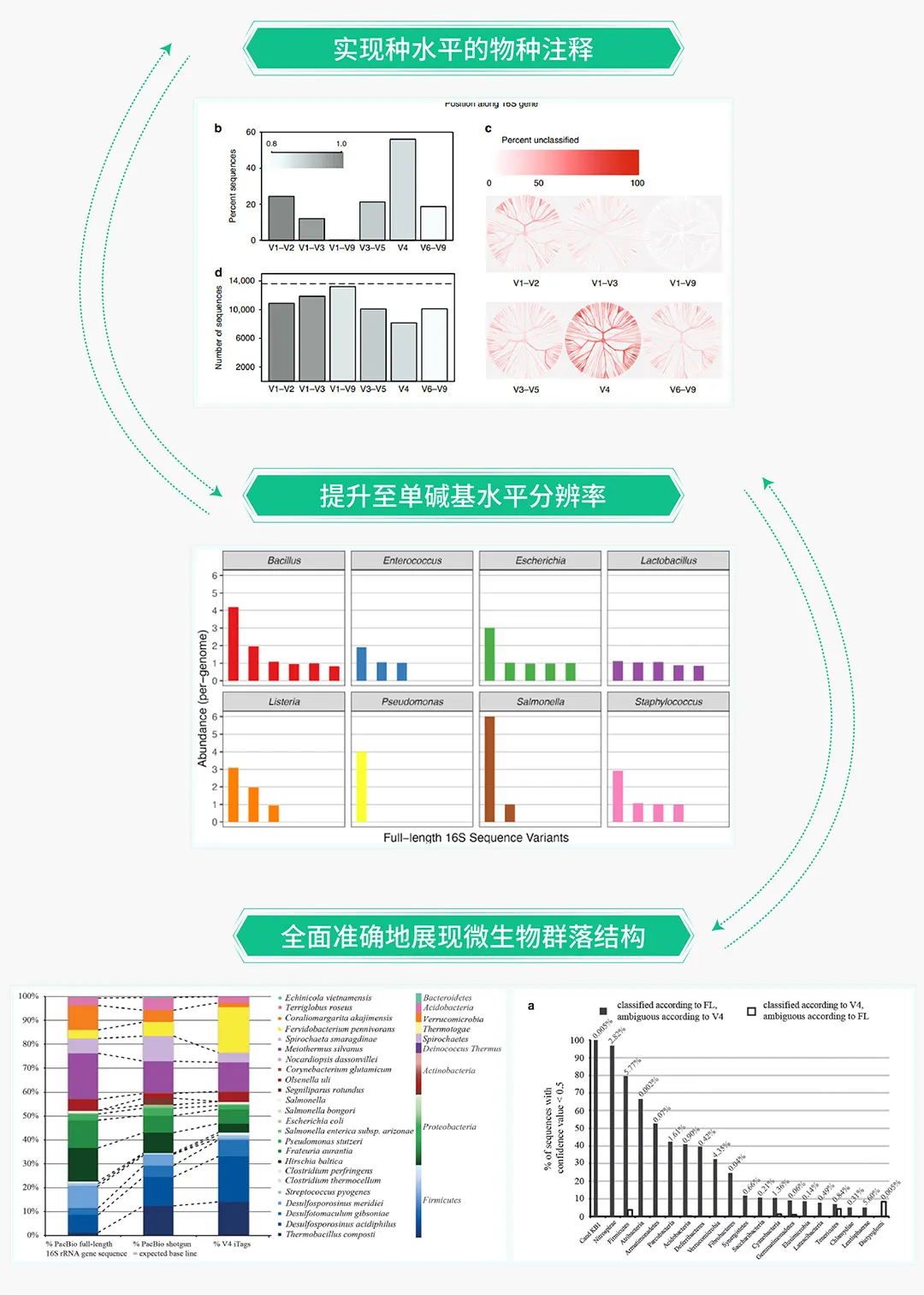
希望组全长16S扩增子全面升级
希望组通过三重技术保障,来提高生产效率、降低生产成本、提高产品精度,从而推动三代扩增子生产的转型升级和高质量交付。
· 自动化生产线设备

希望组通过搭建一整套自动化流程生产线,可实现单日流转样本量>1000个。
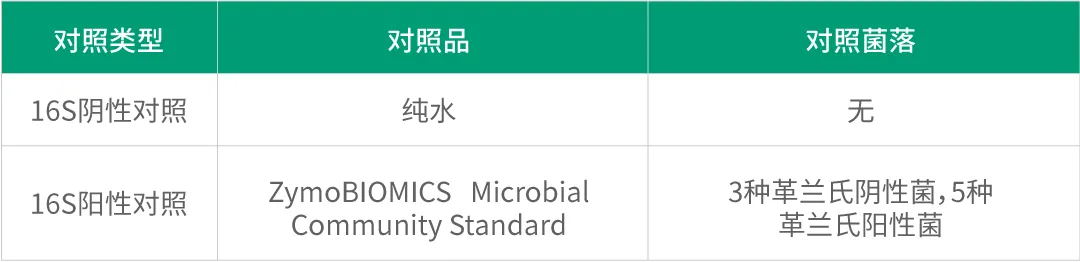
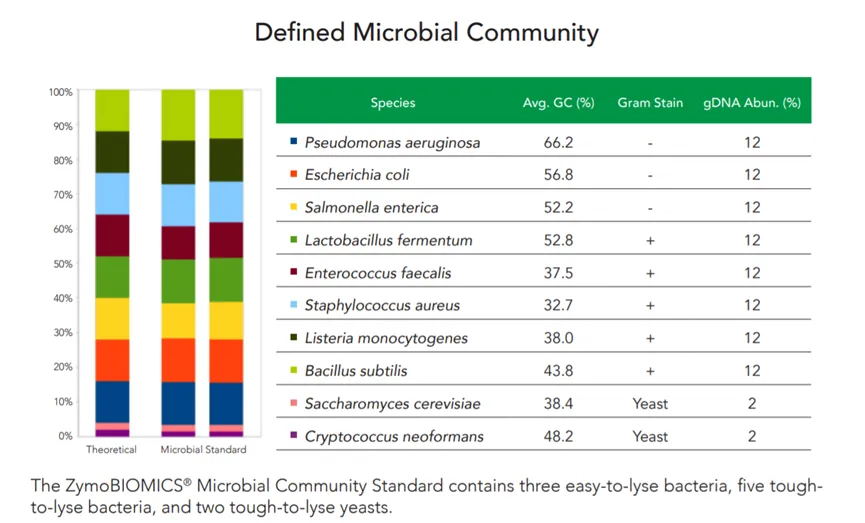
· 阴性和标准品阳性质控
希望组通过每个生产批次独立匹配阴性对照和阳性对照,可以有效地监控实验过程的各个环节,确保数据的准确性和可靠性。其中阴性对照用于检测实验过程中是否存在非特异性扩增或污染,而阳性对照则用于验证实验条件和扩增效率是否正常。
· 串联建库测序流程
PacBio Revio测序平台的全长16S扩增子是基于Kinnex 16S rRNA试剂盒的方法,首先对扩增产物添加特异性barcode序列,将多样本进行混样,可混样的样本数高达384个。kinnex试剂盒还可以将1.5K大小的扩增产物短片段串联成15K以上的长片段,用这种方法构建的文库,可以适配Revio的HiFi测序模式,更具成本效益。
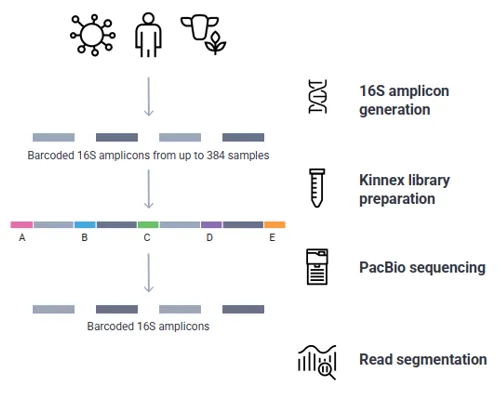
希望组实测数据
· 不同样本类型在不同测序深度条件下检出的物种数量
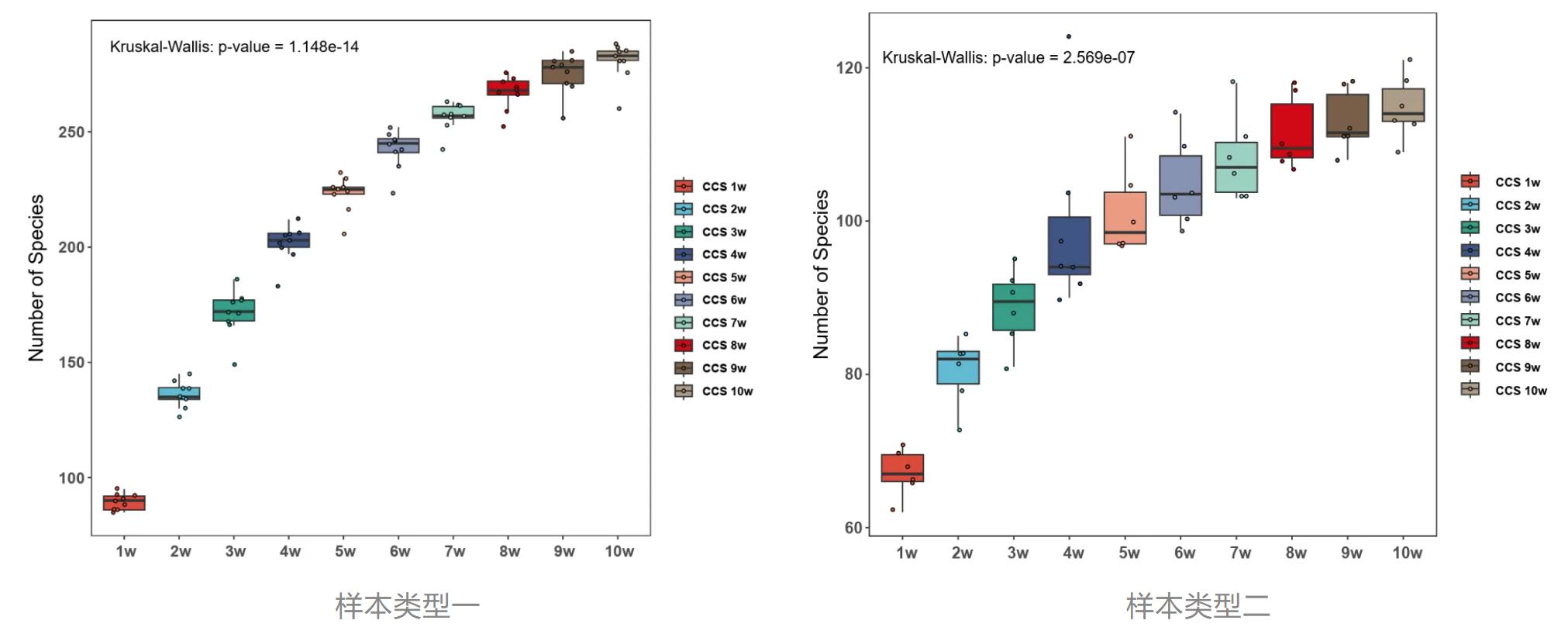
应用方向
三代全长16S测序技术的应用范围极为广泛,在基础科研、环境监测、工农业生产等领域展现其独特的优势!
01.环境检测
题目:Abundant fungi dominate the complexity of microbial networks in soil of contaminated site: High-precision community analysis by full-length sequencing
期刊:Science of The Total Environment
影响因子:IF=10.753
发表时间:2023年2月
样本类型:土壤
该研究对鞍山和台州的6个土壤样本同时进行16S、ITS全长测序以及16S V3-V4、ITS短读长测序,在高分辨率条件下解析受污染土壤中不同微生物类群的群落组成和生态状况。结果表明全长16S rRNA基因测序在所有水平上都能提供更好的细菌鉴定分辨率,在某些样品中的真菌鉴定上没有显著差异。丰富的分类群对于由全长和短读长测序数据构建的微生物共生网络至关重要。上述研究发现有助于了解土壤生态系统中的生态机制和微生物相互作用,并证明全长测序有可能提供更多微生物群落的细节。
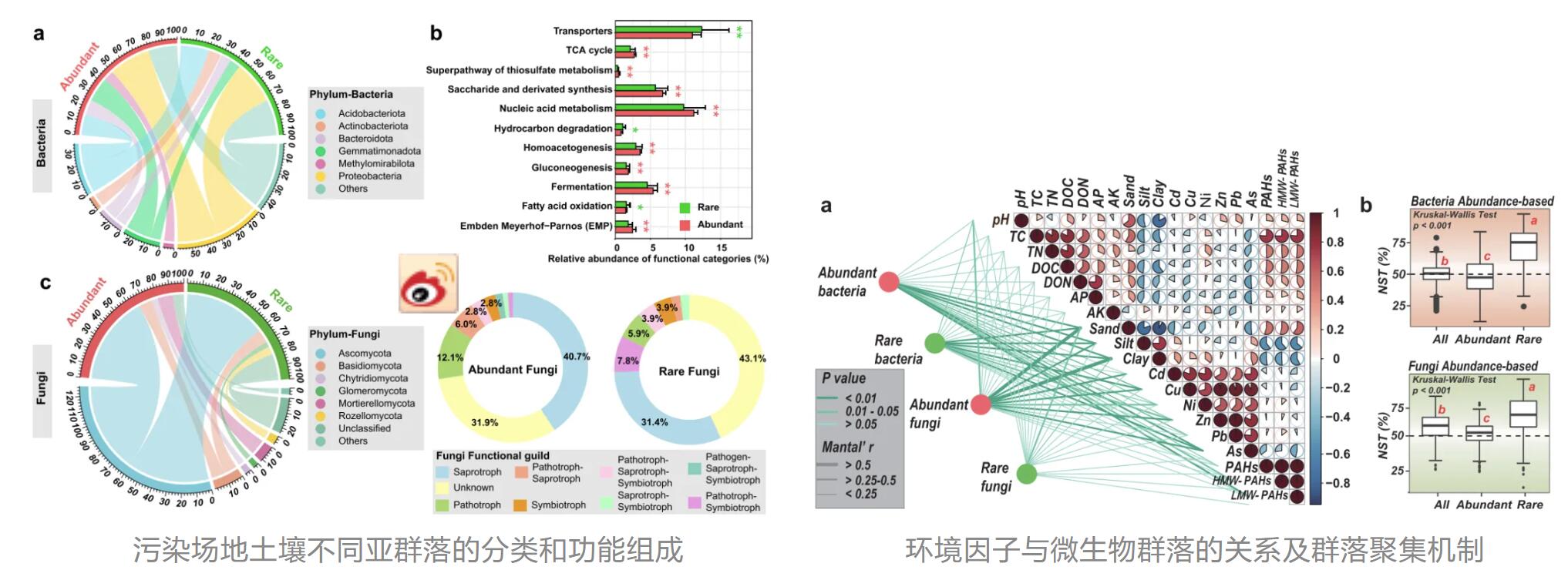
02.工农业生产
期刊:LWT
影响因子:6.056
发表时间:2023年4月
样本类型:发酵产物
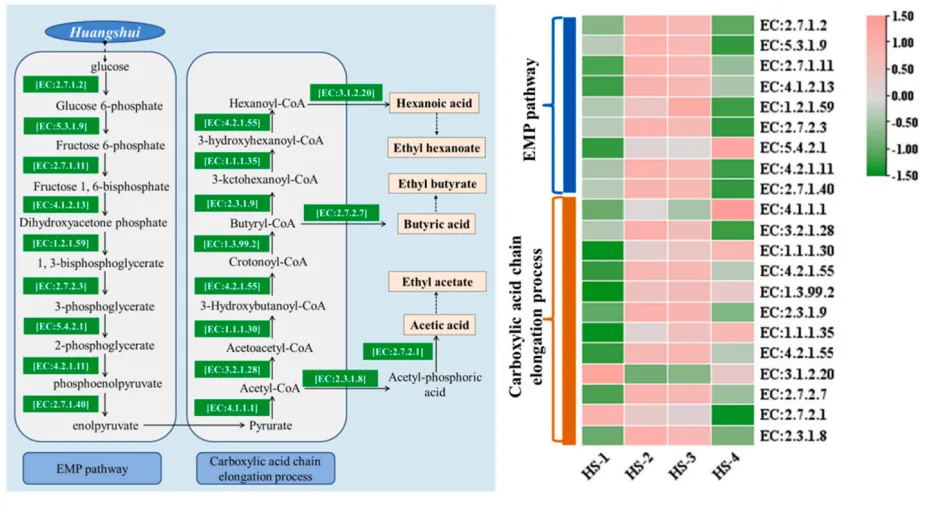
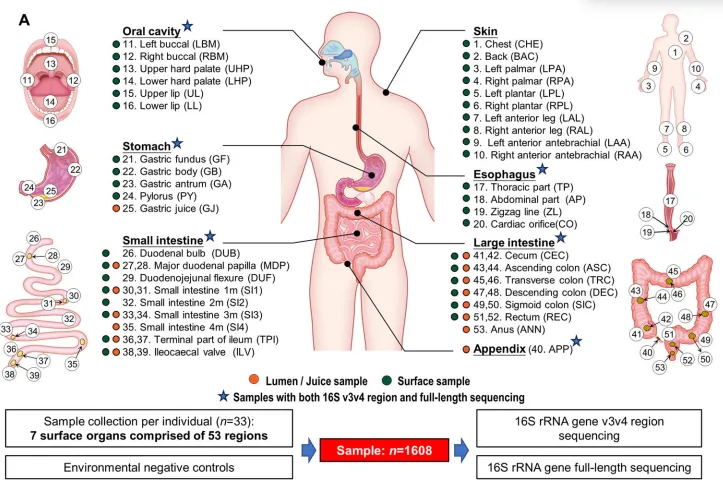
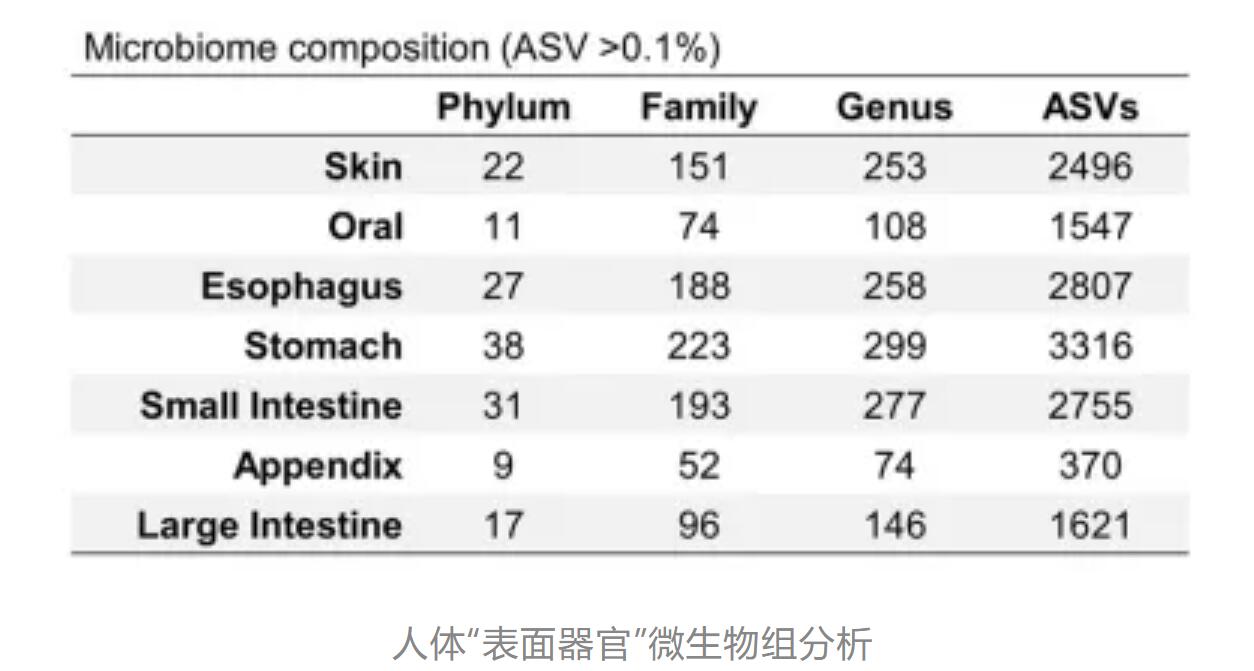
题目:Defining the biogeographical map and potential bacterial translocation of microbiome in human ‘surface organs’
期刊:Nature Communications
影响因子:IF=16.6
发表时间:2024年1月
样本类型:消化系统表面器官(腔和黏膜)和皮肤组织
研究发现与胃、阑尾、小肠或大肠相比,皮肤、口腔和食管的α多样性显著较高,胃的细菌多样性最低,推测其低pH限制了细菌的生长。拟杆菌属狄氏副拟杆菌主要富集在小肠、阑尾和大肠中;卟啉单胞菌属、普雷沃氏菌属、链球菌属和奈瑟菌属富集在口腔中;螺旋菌属在口腔和阑尾中富集;而葡萄球菌属和棒状杆菌属是皮肤中的优势属。
在科学探索的道路上,每一次突破都值得期待。PacBio Revio测序技术以其卓越的性能和可靠性,成为科研领域的璀璨明星。今天,希望组带您直面PacBio Revio 24小时与30小时测序模式的数据对比,从数据产出、reads质量值(QV)、酶读长三个方面揭示其差异与优势。
表1 PacBio Revio 24h VS 30h实测数据对比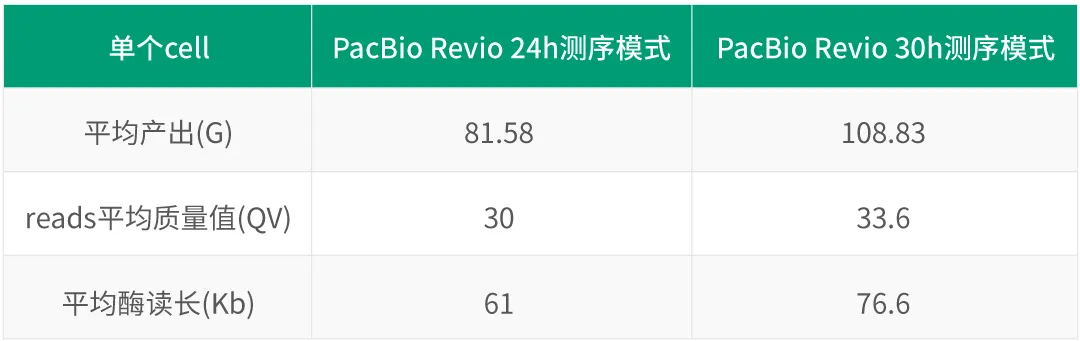
1.数据产出
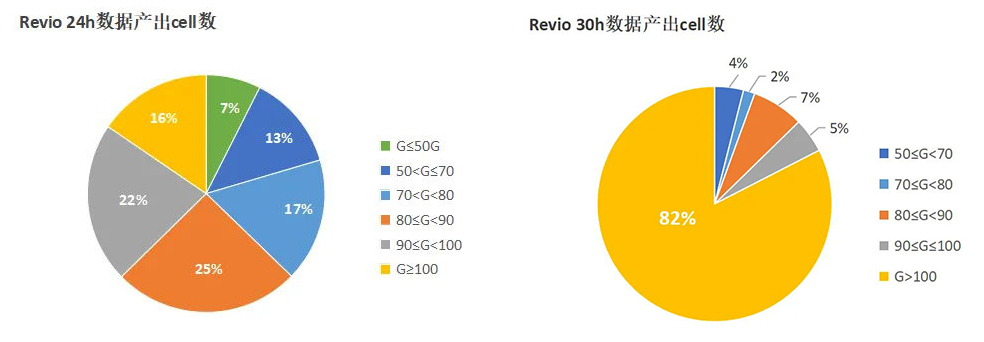
PacBio Revio 24h测序模式单cell平均产出81.58G,30h测序模式单cell平均产出108.83G,提升率超30%。相比之下,30h测序模式带来了单cell更高通量,PacBio Revio单cell数据产出超100G的cell数量占比,由24h 的16%提升至30h的82%,呈现出惊人的提升!
即日起,希望组承诺:质检合格人血液样本PacBio Revio单cell测序数据量不低于100G!
2.reads 质量值
质量值(QV)是衡量测序质量的重要指标,PacBio Revio 30h测序模式展现出更高的单cell平均质量值,相较之下,单cell平均QV值由24h Q30提升至30h Q33.6。PacBio Revio 30h测序模式能够提供更高的测序质量! 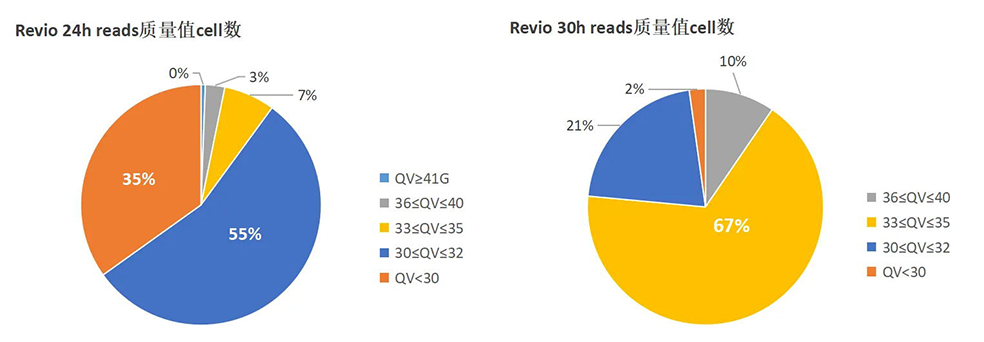
3.酶读长
酶读长直接影响测序结果,PacBio Revio 30h测序模式在酶读长方面表现也相当出色,单cell平均酶读长由24h 61Kb提升至30h 76.6Kb。PacBio Revio 30h测序模式能够为您提供更长、更完整的序列信息,提供更清晰的研究视野! 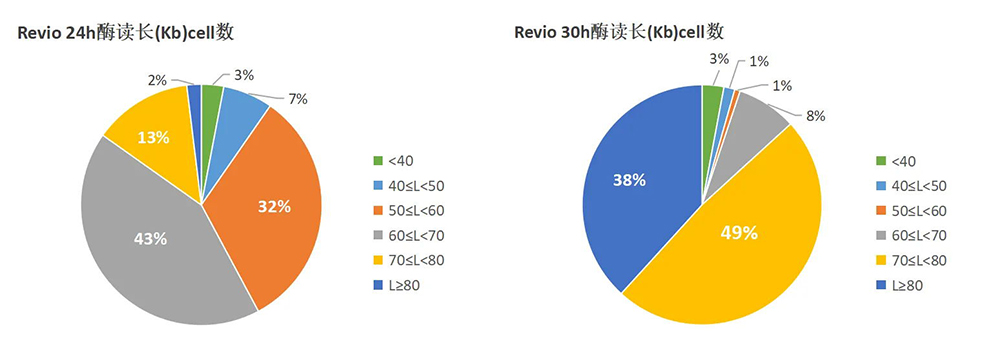
PacBio Revio测序24h VS 30h,在数据产出、reads 质量值、酶读长三方面的对比,30h测序模式表现出绝对的领先优势。即日起,希望组宣布PacBio Revio平台全面升级成30h测序模式!
选择PacBio Revio,选择科研的未来,拥有更多的可能性!让我们携手探索未知,共同揭开基因的奥秘!
近日,Nature Index官网发布了2024自然指数年度榜单(统计自2023年1月1日至2023年12月31日),希望组再度登榜,在大陆的生命科学领域中的测序企业排行榜中名列前茅。排行榜显示,希望组旗下品牌GrandOmics(希望组)和NextOmics(未来组)分别位列第17名和84名;数据合并后,希望组2023年计入自然指数的总Share为0.62,再次进入基因测序行业前3名。
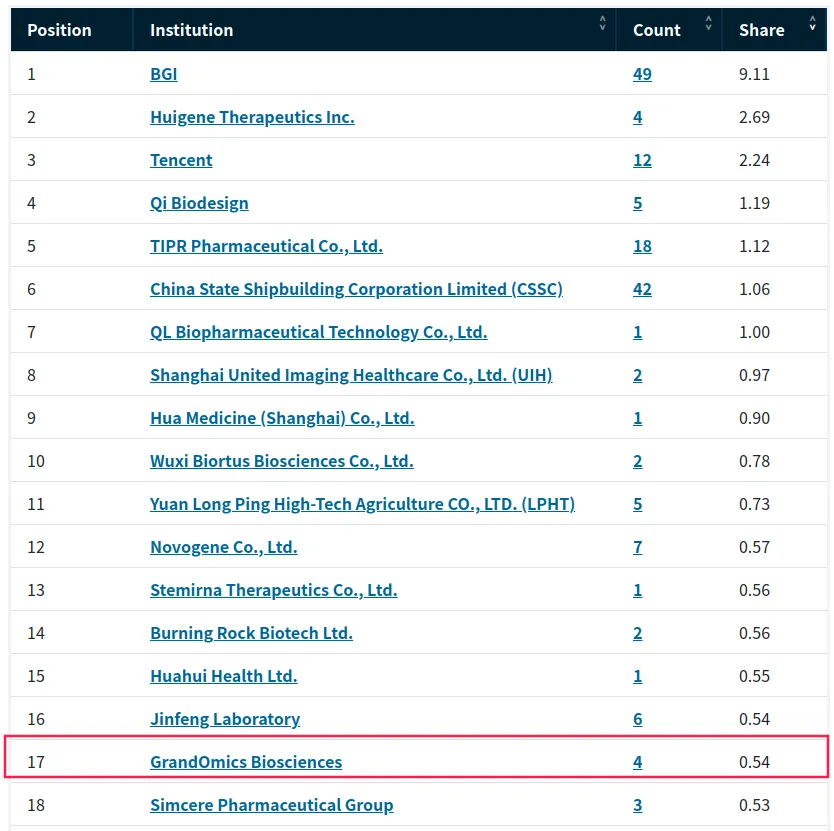

希望组自成立以来一直深耕于长读长测序领域,凭借雄厚的技术积累与合作伙伴的充分信任,在基础科研领域不断有重大科研成果的产出。
2023年希望组共合作发表文章50+篇,总影响因子800+,其中包含Cell、Science、Nature Genetic、Nature Communications等高质量期刊,涵盖基因组、泛基因组、群体基因组、单细胞以及转录组等研究领域。下面让我们一起来回顾一下希望组2023年被收录到自然指数的几篇重要文章吧。
1.Cell :48Gb南极磷虾超大基因组参考序列发布
2023年3月2日,希望组与中国水产科学研究院黄海水产研究所合作在国际顶级期刊Cell (IF=66.85)上发表“The enormous repetitive Antarctic krill genome reveals environmental adaptations and population insights”的研究论文,揭示了南极磷虾适应南大洋的基因组基础,并为未来的南极研究提供了宝贵的资源。研究团队利用PacBio、Hi-C结合短读长对南极磷虾进行测序,使用NextDenovo v2.30 (https://github.com/Nextomics/NextDenovo)组装了48.01Gb的基因组,这是迄今为止报道的最大的动物基因组组装。
(解读链接:署名文章 | Cell!NextDenovo助力破译迄今最大动物基因组—48Gb南极磷虾参考序列)
2.Cell :大规模蛇基因组分析以解析脊椎动物的发育
2023年6月19日,希望组与中国科学院成都生物研究所李家堂团队在Cell (IF=64.5)上发表“Large-scale snake genome analyses provide insights into vertebrate development”的研究论文,该论文基于大规模多组学技术与基因编辑等研究手段,全面揭示了蛇类起源及特有表型演化的遗传机制,对理解脊椎动物演化历史具有重要意义。
(解读链接:项目文章 | Cell!李家堂团队揭示蛇类的起源与演化机制)
3.Science :系统基因组学分析对灵长类演化进程提供见解
2023年6月2日,希望组与浙江大学生命演化研究中心张国捷教授团队联合昆明动物研究所吴东东教授团队、西北大学齐晓光教授团队等在Science (IF=56.9)上发表“Phylogenomic analyses provide insights into primate evolution”的研究论文,该研究对14科38属的50个灵长类物种进行分析,揭示了基因组重排和基因进化的异质性,发现不同谱系中处于正向选择下的数千个基因在神经、骨骼和消化系统中发挥作用。该研究还揭示了许多关键的基因组变异发生在类人猿下目祖先节点,并且可能对其适应性辐射和人类的进化产生影响。
4.Nature Genetics :玉米T2T基因组组装
2023年6月15日,希望组与中国农业大学国家玉米改良中心、玉米生物育种全国重点实验室赖锦盛教授团队以题为“A complete telomere-to-telomere assembly of the maize genome”在Nature Genetics(IF=30.8)上在线发表了玉米全基因组所有染色体端粒到端粒完整无间隙组装结果,在复杂动植物基因组中第一个实现真正意义上的全基因组完整无间隙组装。该研究是复杂基因组组装领域工程技术研究的重大突破,攻克了复杂动植物基因组组装的最后一道难题,是基因组组装和基因组学研究的一个重要里程碑。
(解读链接:署名文章 | Nature Genetics!希望组携手赖锦盛教授团队再创新里程—大型真核生物玉米T2T无间隙基因组)
ONT平台的测序可产生>100 Kb的超长reads用于填充基因组组装中串联或高度同源的多拷贝重复区域,但其同时伴随着准确度不高的问题。使用ONT数据组装基因组,有两种常使用策略即“先矫正后组装”(CTA)和“先组装后矫正”(ATC),对于大型植物基因组在组装重复序列时,基于CTA策略通常能产生更准确和连续的组装。对此,希望组自主研发基于ONT数据进行高效纠错和CTA组装的NextDenovo 软件,用于组装出一个完整、准确的基因组。NextDenovo 软件历经多年打磨以及在动植物基因组组装中的成功应用,于2024年4月26日在《Genome Biology》发表题为《NextDenovo:an efficient error correction and accurate assembly tool for noisy long reads》的文章。

NextDenovo 包含五个主要步骤:1、对原始reads进行成对重叠;2、过滤重叠结果避免错误配对以影响纠错的准确性;3、基于过滤后的重叠结果进行纠错;4、需要两步迭代成对矫正reads重叠;5、使用重叠结果构建一个组装图,然后进行图形清理并输出结果。 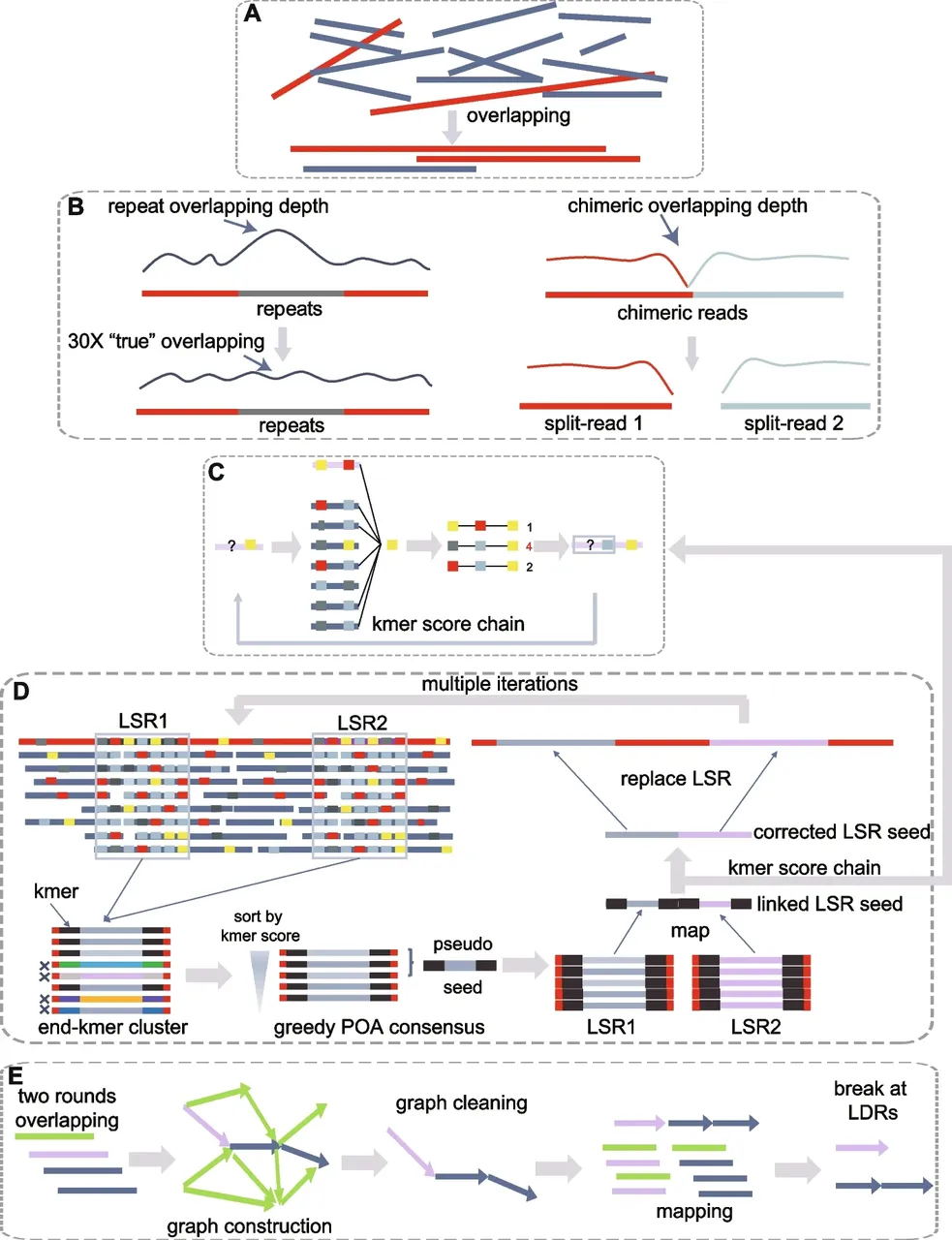
图1. NextDenovo 组装流程图
在纠错速度方面,NextDenovo与 Consent、Canu 和 Necat 相比,在模拟数据上分别快 3.00 倍、7.44 倍和 1.13 倍;在真实数据上则分别快 9.51 倍、69.25 倍和 1.63 倍。
表1. ONT reads纠错统计 
将NextDenovo软件与其他纠错、组装软件在4个非人类基因组(果蝇、拟南芥、水稻、玉米)和35个人类基因组的组装方面进行了比较,结果显示NextDenovo可快速、高效地对ONT数据进行纠错并产生高准确度的基因组组装,特别是对于含有大量重复序列的基因组。 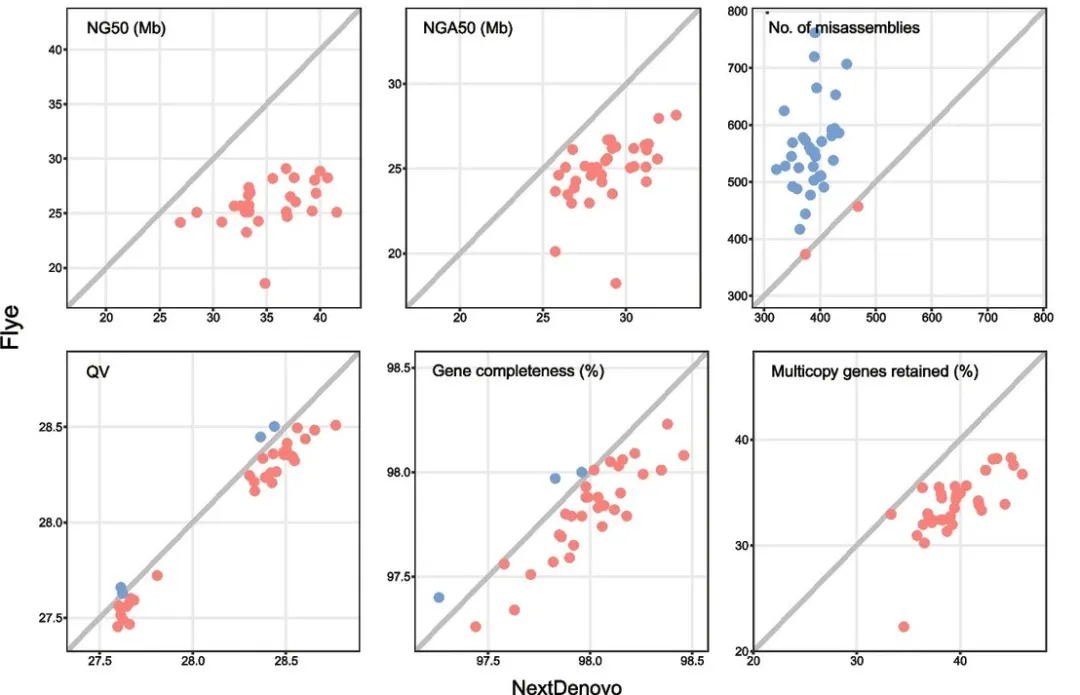
图2. 35个人类基因组的De novo组装
NextDenovo 已成功多次用于大基因组组装,例如约 10.5 Gb 的苏铁(Cycas panzhihuaensis)基因组组装(contig N50 = 12 Mb)、约 10.76 Gb 的六倍体燕麦基因组组装(contig N50 = 75.27 Mb)、约 40 Gb 的非洲肺鱼基因组组装(contig N50 = 1.60 Mb)和约 48 Gb 的南极磷虾基因组组装(contig N50 = 178.99 kb)。
通过使用 ONT超长reads,NextDenovo 可以产生部分或几乎达到染色体水平的组装。在约 4.59 Gb 罂粟基因组中,NextDenovo 使用约 19X ONT超长reads和约 86X ONT常规reads组装了 contig N50 为 65.57 Mb的基因组,最长长度为 178.776 Mb ;类似地,对于 3.69 Gb 的西瓜基因组,NextDenovo 使用约 57X ONT超长reads,组装出11 条最长 contig 表示 11 条染色体;在约 10.76 Gb 的六倍体燕麦基因组中,NextDenovo 使用约 100X ONT超长reads 组装了contig N50 为 75.27 Mb,最长长度为 313.87 Mb的基因组。
总的来说,NextDenovo 是一种针对长读长的高效纠错和组装工具,它可以快速提供高度准确的纠错reads,并从这些reads中产生准确的组装。特别是当使用 ONT 的超长reads进行组装时,NextDenovo 可以生成部分或接近染色体级的组装。ONT测序具有低成本、高通量、周期快的特点,因此NextDenovo 还是一种用于群体规模的ONT长读长测序数据的优秀组装工具。
希望组一直致力于自主创新、开发优质软件以便为客户交付更优质的高质量数据用于后续的科学研究,以助力各位专家学者在基因组学领域取得更多的突破和进展!除NextDenovo外,希望组自研软件NextPolish可高效矫正三代(Nanopore 、Pacbio)下机数据组装得到基因组的单碱基错误,进一步提高单碱基准确性。该工具采用 K-mer 得分链和 K-mer 计数算法,在运行速度、校正精度及消耗资源等方面均优于同类软件,NextPolish 目前已在《Bioinformatics》 期刊正式发表《NextPolish: a fast and efficient genome polishing tool for long-read assembly》。

希望组

希望组科技服务

希望组诊断服务